A recent work published in eLife (Hu et al. DNA methylation presents distinct binding sites for human transcription factors. eLife 2013) challenges the notion of suppressive methylated CpG islands by reporting a significant number of transcription factors binding preferentially to methylated cytosines of CpG islands. In mammals, the methylation of CpG sites—which consist of a cytosine base next to a guanine base—is typically thought to reduce gene expression by preventing proteins called transcription factors from binding to regions of DNA called promoters. This can occur directly if methylation disrupts interactions between the DNA and the transcription factors, or indirectly if other proteins that bind to the methylated DNA compete with the transcription factors for binding sites. However, only a small number of proteins that bind to methylated DNA have so far been identified.
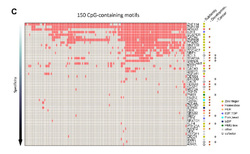
The data used: The authors use protein arrays for 1300 TF and their co-factors in order to assess their binding affinity on unmethylated or methylated DNA. The DNA stretches used were in total 154 sequences selected on the basis of high probability to form part of human promoters, being representative of known TF-binding sites and carrying at least one CpG site. TF-binding intensities were then measured for both the unmethylated and the methylated version of each of the sequences.
The analysis: Differential TF binding for methylated and unmethylated revealed a significant subset (47 proteins) showed increased binding for the CpG-methylated DNA instead of the unmethylated one. The authors showed that this represents an inherent property of the proteins by showing selective binding of specific TF towards different DNA sequences when they contain methylated cytosines and when not. In this sense, the authors coin mC (methylcytosine) as the "fifth base".
What's next: As the authors note the number of TF identified in this study is probably an under-estimation since only a very limited number of DNA targets was used. High-throughput techniques coupling of high-resolution DNA methylation (RRBS) with ChIPSeq to define regions of TF binding that are effectively methylated are probably the most effective way to probe methylated DNA binding directly.
Read more: A work published a bit earlier (Spruijt et al. Cell 2012) where specific direct binding of hydroxy-methylcytosines is assayed at genome-scale. hmC (hydroxy-methylcytosine) is probably the primary candidate for being coined as the "sixth base".

 RSS Feed
RSS Feed
