Two recent works, one published in Plos One (Park et al. Widespread misinterpretable ChIPSeq bias in yeast) and one in PNAS (Teytelman et al. Highly expressed loci are vulnerable to misleading ChIPlocalization of multiple unrelated proteins)
point out a very important artifact of a very widely applied method, ChIPSeq. Both papers that appeared almost simultaneously show that there is an inherent bias in all ChIP experiments to result in enriching regions belonging to highly-expressed genes regardless of the nature of the studied protein.
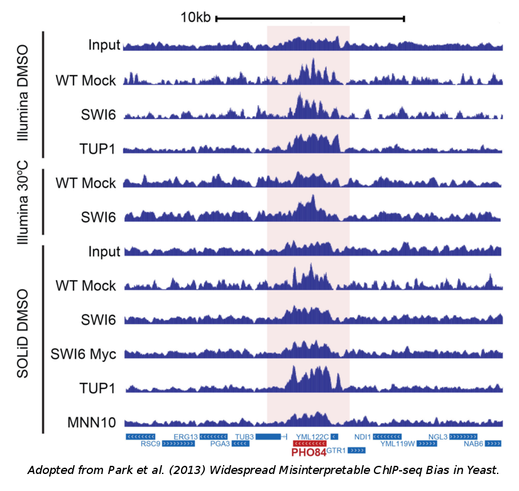
The data used: Both groups performed a series of experiments in yeast but their results may readily be extended to more complex genomes. Park et al. mostly focused on Tup1, a well-studied transcriptional supressor, the binding of which they surprisingly found to coincide with highly-expressed genes under multiple conditions. Teytelman et al. did the same starting from a family of repressors known as Sir proteins which they also saw to be consistently located in genes with high-expression.
The analysis: The analysis was pretty straight-forward. ChIPSeq data from various platforms were analyzed with standard peak-calling algorithms, such as MACS2 and the discovered peaks were correlated with the expression of the closest or overlapping genes. In all cases, a positive correlation was observed even if the protein under study was not a transcriptional activator. Teytelman et al. were able to show that a positive corrrelation was even observed in the case of a GFP ChIP experiment, that is a "mock" experiment with a protein that does not bind the DNA under normal conditions. The authors, thus, reach the conclusion that the major source of bias is the process of cross-linking which is likely to occur preferentially in actively transcribed regions.
What's next: Both papers point out a very important aspect that has largely escaped the attention of a huge number of published works. At the same time both groups suggest the performance of extensive controls for every ChIPSeq experiment to be performed in the future. For instance, performing a "mock" ChIP with a non-DNA-binding protein such as GFP and subtracting the signal (as representative of hyper-chipability) would distinguish signal from noise. This is bound to work to a great extent but is probably not cost-worthy, while it will complicate the downstream analysis through the addition of one more condition. On the other hand, new approaches that do away with cross-linking may provide more effective solutions.
Read more: A work published this week in Nature Methods by the group of S. Henikoff describing a methodology for inquiring transcription factor binding in native (non cross-linked) chromatin (Kazinathan et al. High-resolution mapping of transcription factor binding sites on native chromatin). This is probably the next step in this sort of approaches as native chromatin does away with cross-linking which is the main source of highly-expressed gene bias.
point out a very important artifact of a very widely applied method, ChIPSeq. Both papers that appeared almost simultaneously show that there is an inherent bias in all ChIP experiments to result in enriching regions belonging to highly-expressed genes regardless of the nature of the studied protein.
The data used: Both groups performed a series of experiments in yeast but their results may readily be extended to more complex genomes. Park et al. mostly focused on Tup1, a well-studied transcriptional supressor, the binding of which they surprisingly found to coincide with highly-expressed genes under multiple conditions. Teytelman et al. did the same starting from a family of repressors known as Sir proteins which they also saw to be consistently located in genes with high-expression.
The analysis: The analysis was pretty straight-forward. ChIPSeq data from various platforms were analyzed with standard peak-calling algorithms, such as MACS2 and the discovered peaks were correlated with the expression of the closest or overlapping genes. In all cases, a positive correlation was observed even if the protein under study was not a transcriptional activator. Teytelman et al. were able to show that a positive corrrelation was even observed in the case of a GFP ChIP experiment, that is a "mock" experiment with a protein that does not bind the DNA under normal conditions. The authors, thus, reach the conclusion that the major source of bias is the process of cross-linking which is likely to occur preferentially in actively transcribed regions.
What's next: Both papers point out a very important aspect that has largely escaped the attention of a huge number of published works. At the same time both groups suggest the performance of extensive controls for every ChIPSeq experiment to be performed in the future. For instance, performing a "mock" ChIP with a non-DNA-binding protein such as GFP and subtracting the signal (as representative of hyper-chipability) would distinguish signal from noise. This is bound to work to a great extent but is probably not cost-worthy, while it will complicate the downstream analysis through the addition of one more condition. On the other hand, new approaches that do away with cross-linking may provide more effective solutions.
Read more: A work published this week in Nature Methods by the group of S. Henikoff describing a methodology for inquiring transcription factor binding in native (non cross-linked) chromatin (Kazinathan et al. High-resolution mapping of transcription factor binding sites on native chromatin). This is probably the next step in this sort of approaches as native chromatin does away with cross-linking which is the main source of highly-expressed gene bias.
 RSS Feed
RSS Feed
