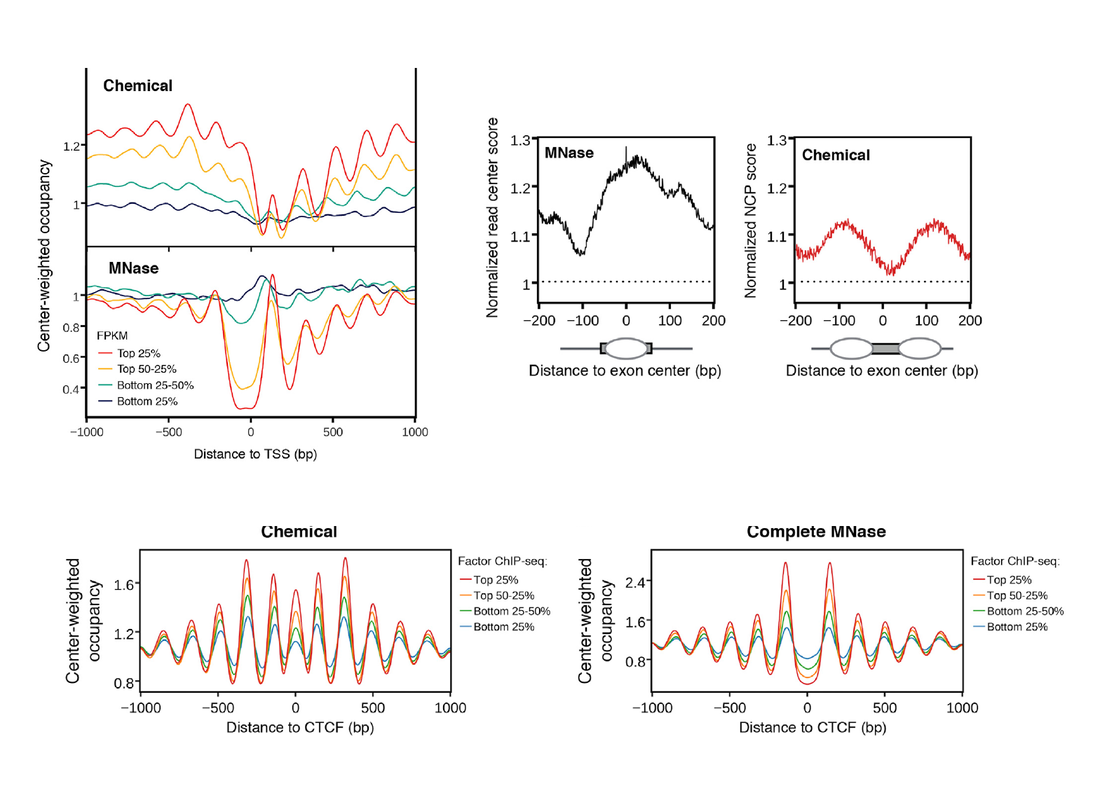
Just the other day we were discussing a new chemical-coupled NGS method that threatened to change our view on nucleosome positioning, according to which a great proportion of nucleosomes which are simply absent from MNase digestion maps but are revealed to occupy regions where once we thought they were not supposed to.
In a recent paper in Genome Research, Tess Jeffers and Jason Lieb, bring back the notion of fragile nucleosomes, this time through conventional "good-old" MNase-Seq. In a nutshell, what Jeffers and Lieb did is not very different from early works on the concept of fragile nucleosomes (i.e. nucleosomes that can be digested by MNase and are thus "lost" from MNase maps) in the sense that they use differential timing MNase digestion, separating the output and treating a specific set of sequences sizes that comes from low digestion times as the "fragile" fraction.
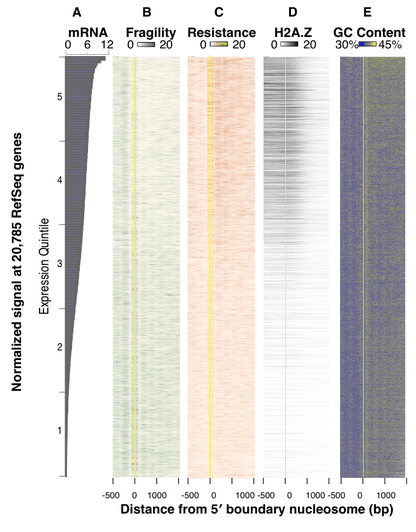
Through a rather straightforward analysis of differential nucleosome positioning in C. elegans embryos, the authors were able to recap most of what we knew about nucleosomes already. Fragile nucleosomes are AT-rich, enriched in promoters of low-expression genes, lacking enrichments in regulatory activating marks and isoforms such as H2A.Z. This reassuring(?) image is summarized in the Figure below (Figure 4a in their paper), where fragility and resistance correspond to the fragile and "well-positioned" fractions of the bulk nucleosomes.
In a recent paper in Genome Research, Tess Jeffers and Jason Lieb, bring back the notion of fragile nucleosomes, this time through conventional "good-old" MNase-Seq. In a nutshell, what Jeffers and Lieb did is not very different from early works on the concept of fragile nucleosomes (i.e. nucleosomes that can be digested by MNase and are thus "lost" from MNase maps) in the sense that they use differential timing MNase digestion, separating the output and treating a specific set of sequences sizes that comes from low digestion times as the "fragile" fraction.
Through a rather straightforward analysis of differential nucleosome positioning in C. elegans embryos, the authors were able to recap most of what we knew about nucleosomes already. Fragile nucleosomes are AT-rich, enriched in promoters of low-expression genes, lacking enrichments in regulatory activating marks and isoforms such as H2A.Z. This reassuring(?) image is summarized in the Figure below (Figure 4a in their paper), where fragility and resistance correspond to the fragile and "well-positioned" fractions of the bulk nucleosomes.
A number of aspects touched upon the paper of Voong et al., such as exon-intron nucleosomes (a matter of personal interest) or different promoter classes remain unchallenged by this study. One however is and in a particularly informative way. Jeffers and Lieb show that fragile nucleosomes are extremely enriched in areas of the genome where a number of transcription factors are expected to bind, which can explain the discrepancy in previous maps of MNase-defined nucleosomes and pioneer-factors or CTCF binding sites.
Still, as this little story develops it would be interesting to see if the chemical-mapping method was just a firework or is here to make a difference.
Still, as this little story develops it would be interesting to see if the chemical-mapping method was just a firework or is here to make a difference.
 RSS Feed
RSS Feed
