Scientific Papers |
On genome architecture and evolution
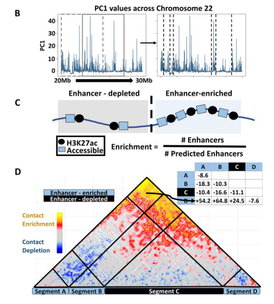
SEGCOND predicts putative transcriptional condensate-associated genomic regions by integrating multi-omics data
Antonios Klonizakis, Christoforos Nikolaou and Thomas Graf. (2023) Bioinformatics, 39,1: btac742 (https://doi.org/10.1093/bioinformatics/btac742)
In a nutshell:
The compartmentalization of biochemical reactions, involved in the activation of gene expression in the
eukaryotic nucleus, leads to the formation of membraneless bodies through liquid–liquid phase separation. These formations, called transcriptional condensates, appear to play important roles in gene regulation as they are assembled through the association of multiple enhancer regions in 3D genomic space. To date, we are still lacking efficient computational methodologies to identify the regions responsible for the formation of such condensates, based on genomic and conformational data.
In this work, we present SEGCOND, a computational framework aiming to highlight genomic regions
involved in the formation of transcriptional condensates. SEGCOND is flexible in combining multiple genomic
datasets related to enhancer activity and chromatin accessibility, to perform a genome segmentation. It then uses this segmentation for the detection of highly transcriptionally active regions of the genome. At a final step, and through the integration of Hi-C data, it identifies regions of putative transcriptional condensates (PTCs) as genomic domains where multiple enhancer elements coalesce in 3D space. SEGCOND identifies a subset of enhancer segments with increased transcriptional activity. PTCs are also found to significantly overlap highly interconnected enhancer elements and super enhancers obtained through two independent approaches. Application of SEGCOND on data from a well-defined system of B-cell to macrophage transdifferentiation leads to the identification of previously unreported genes with a likely role in the process.
Antonios Klonizakis, Christoforos Nikolaou and Thomas Graf. (2023) Bioinformatics, 39,1: btac742 (https://doi.org/10.1093/bioinformatics/btac742)
In a nutshell:
The compartmentalization of biochemical reactions, involved in the activation of gene expression in the
eukaryotic nucleus, leads to the formation of membraneless bodies through liquid–liquid phase separation. These formations, called transcriptional condensates, appear to play important roles in gene regulation as they are assembled through the association of multiple enhancer regions in 3D genomic space. To date, we are still lacking efficient computational methodologies to identify the regions responsible for the formation of such condensates, based on genomic and conformational data.
In this work, we present SEGCOND, a computational framework aiming to highlight genomic regions
involved in the formation of transcriptional condensates. SEGCOND is flexible in combining multiple genomic
datasets related to enhancer activity and chromatin accessibility, to perform a genome segmentation. It then uses this segmentation for the detection of highly transcriptionally active regions of the genome. At a final step, and through the integration of Hi-C data, it identifies regions of putative transcriptional condensates (PTCs) as genomic domains where multiple enhancer elements coalesce in 3D space. SEGCOND identifies a subset of enhancer segments with increased transcriptional activity. PTCs are also found to significantly overlap highly interconnected enhancer elements and super enhancers obtained through two independent approaches. Application of SEGCOND on data from a well-defined system of B-cell to macrophage transdifferentiation leads to the identification of previously unreported genes with a likely role in the process.
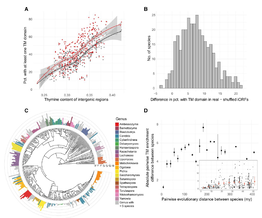
Intergenic Regions of Saccharomycotina Yeasts are Enriched in Potential to Encode Transmembrane Domains.
Emilios Tassios, Christoforos Nikolaou and Nikolaos Vakirlis (2023). Molecular Biology and Evolution, 40 (3): msad059 ( https://doi.org/10.1093/molbev/msad059)
In a nutshell:
Ever wondered if genes can emerge de novo from previously non-coding sequences? Well, it turns out that this happens more often than one would expect and that, in fact, de novo gene emergence is an until recently overlooked process of gene innovation in all species, especially in the gene-sparse genomes of eukaryotes. Starting from a set of >300 aligned fungal genomes, in this work we compiled the largest -to date- compendium of bona fide de novo emerged genes and went on to study one particular, very interesting property, namely the tendency of these "young" coding sequences to encode for transmembrane (TM) domains. We find pervasive but variable enrichment in TM-form ing potential across the subphylum regardless of the composition and average size of intergenic regions. This cryptic property is evenly spread across the genome, cannot be explained by the hydrophobic content of the sequence, and does not appear to localize to regions containing regulatory motifs. This TM-forming enrichment specifically, and not the actual TM-forming potential, is associated, across genomes, with more TM domains in evolutionarily young genes. Our findings shed light on this newly discovered feature of yeast genomes and constitute a first step toward
understanding its evolutionary importance.
Emilios Tassios, Christoforos Nikolaou and Nikolaos Vakirlis (2023). Molecular Biology and Evolution, 40 (3): msad059 ( https://doi.org/10.1093/molbev/msad059)
In a nutshell:
Ever wondered if genes can emerge de novo from previously non-coding sequences? Well, it turns out that this happens more often than one would expect and that, in fact, de novo gene emergence is an until recently overlooked process of gene innovation in all species, especially in the gene-sparse genomes of eukaryotes. Starting from a set of >300 aligned fungal genomes, in this work we compiled the largest -to date- compendium of bona fide de novo emerged genes and went on to study one particular, very interesting property, namely the tendency of these "young" coding sequences to encode for transmembrane (TM) domains. We find pervasive but variable enrichment in TM-form ing potential across the subphylum regardless of the composition and average size of intergenic regions. This cryptic property is evenly spread across the genome, cannot be explained by the hydrophobic content of the sequence, and does not appear to localize to regions containing regulatory motifs. This TM-forming enrichment specifically, and not the actual TM-forming potential, is associated, across genomes, with more TM domains in evolutionarily young genes. Our findings shed light on this newly discovered feature of yeast genomes and constitute a first step toward
understanding its evolutionary importance.
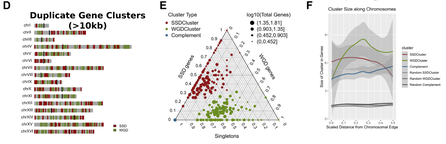
Distinct chromosomal “niches” in the genome of Saccharomyces cerevisiae provide the background for genomic innovation and shape the fate of gene duplicates
Athanasia Stavropoulou, Emilios Tassios, Maria Kalyva, Michalis Georgoulopoulos, Nikolaos Vakirlis, Ioannis Iliopoulos and Christoforos Nikolaou (2022), Nucleic Acids Research - Genomics and Bioinformatics, 4, 4, 1-12
In a nutshell:
Nearly one third of Saccharomyces cerevisiae protein coding sequences correspond to duplicate genes, equally split between small-scale duplicates (SSD) and whole-genome duplicates (WGD). While duplicate genes have distinct properties compared to singletons, to date, there has been no systematic analysis of their positional preferences. In this work, we show that SSD and WGD genes are organized in distinct gene clusters that occupy different genomic regions, with SSD being more peripheral and WGD more centrally positioned close to centromeric chromatin. Duplicate gene clusters differ from the rest of the genome in terms of gene size and spacing, gene expression variability and regulatory complexity, properties that are also shared by singleton genes residing within them. Singletons within duplicate gene clusters have longer promoters, more complex structure and a higher number of protein–protein interactions. Particular chromatin architectures appear to be important for gene evolution, as we find SSD gene-pair co-expression to be strongly associated with the similarity of nucleosome positioning patterns. We propose that specific regions of the yeast genome provide a favourable environment for the generation and maintenance of small-scale gene duplicates, segregating them from WGD-enriched genomic domains. Our findings provide a valuable framework linking genomic innovation with positional genomic preferences.
You can read more about this in this twitter thread: https://twitter.com/guilderstern/status/1498964712662474753
Athanasia Stavropoulou, Emilios Tassios, Maria Kalyva, Michalis Georgoulopoulos, Nikolaos Vakirlis, Ioannis Iliopoulos and Christoforos Nikolaou (2022), Nucleic Acids Research - Genomics and Bioinformatics, 4, 4, 1-12
In a nutshell:
Nearly one third of Saccharomyces cerevisiae protein coding sequences correspond to duplicate genes, equally split between small-scale duplicates (SSD) and whole-genome duplicates (WGD). While duplicate genes have distinct properties compared to singletons, to date, there has been no systematic analysis of their positional preferences. In this work, we show that SSD and WGD genes are organized in distinct gene clusters that occupy different genomic regions, with SSD being more peripheral and WGD more centrally positioned close to centromeric chromatin. Duplicate gene clusters differ from the rest of the genome in terms of gene size and spacing, gene expression variability and regulatory complexity, properties that are also shared by singleton genes residing within them. Singletons within duplicate gene clusters have longer promoters, more complex structure and a higher number of protein–protein interactions. Particular chromatin architectures appear to be important for gene evolution, as we find SSD gene-pair co-expression to be strongly associated with the similarity of nucleosome positioning patterns. We propose that specific regions of the yeast genome provide a favourable environment for the generation and maintenance of small-scale gene duplicates, segregating them from WGD-enriched genomic domains. Our findings provide a valuable framework linking genomic innovation with positional genomic preferences.
You can read more about this in this twitter thread: https://twitter.com/guilderstern/status/1498964712662474753
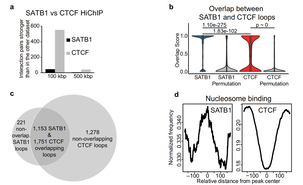
The 3D enhancer network of the developing T cell genome is shaped by SATB1
(in collaboration with the lab of Babis Spilianakis, University of Crete and IMBB-FORTH)
Tomas Zelenka, Antonios Klonizakis, Despina Tsoukatou, Dionysios-Alexandros Papamatheakis, Sören Franzenburg, Petros Tzerpos, Ioannis-Rafail Tzonevrakis, George Papadogkonas, Manouela Kapsetaki, Christoforos Nikolaou, Dariusz Plewczynski, Charalampos Spilianakis
(2022) Nature Communications, 13, 1, 1-22
In a nutshell:
SATB1 is a protein required for the maturation of T-cells in the thymus. Until recently, little was known of its role, other than that it binds extensively on the DNA with a preference for AT-rich sequences near numerous genes. In this work we have conducted a thorough analysis of SATB1 binding in T-cells through a conditional knock-out mouse model. We integrated SATB1 binding patterns with chromosomal organization through Hi-ChIP and Hi-C experiments and with gene activation via H3K27ac. We found that SATB1 plays a key role in the establishment and maintenance of enhancer-promoter and enhancer-enhancer interactions for genes that are specific for CD4/CD8 double positive T-cells. SATB1-dependent regulatory chromatin loops represent a more refined layer of genome organization built upon a high-order scaffold provided by CTCF and other factors. Overall, our findings unravel the function of a tissue-specific factor that controls transcription programs, via spatial chromatin arrangements complementary to the chromatin structure imposed by ubiquitously expressed genome organizers.
(in collaboration with the lab of Babis Spilianakis, University of Crete and IMBB-FORTH)
Tomas Zelenka, Antonios Klonizakis, Despina Tsoukatou, Dionysios-Alexandros Papamatheakis, Sören Franzenburg, Petros Tzerpos, Ioannis-Rafail Tzonevrakis, George Papadogkonas, Manouela Kapsetaki, Christoforos Nikolaou, Dariusz Plewczynski, Charalampos Spilianakis
(2022) Nature Communications, 13, 1, 1-22
In a nutshell:
SATB1 is a protein required for the maturation of T-cells in the thymus. Until recently, little was known of its role, other than that it binds extensively on the DNA with a preference for AT-rich sequences near numerous genes. In this work we have conducted a thorough analysis of SATB1 binding in T-cells through a conditional knock-out mouse model. We integrated SATB1 binding patterns with chromosomal organization through Hi-ChIP and Hi-C experiments and with gene activation via H3K27ac. We found that SATB1 plays a key role in the establishment and maintenance of enhancer-promoter and enhancer-enhancer interactions for genes that are specific for CD4/CD8 double positive T-cells. SATB1-dependent regulatory chromatin loops represent a more refined layer of genome organization built upon a high-order scaffold provided by CTCF and other factors. Overall, our findings unravel the function of a tissue-specific factor that controls transcription programs, via spatial chromatin arrangements complementary to the chromatin structure imposed by ubiquitously expressed genome organizers.
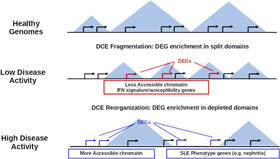
Extensive fragmentation and re-organization of transcription in Systemic Lupus Erythematosus
(in collaboration with the lab of George Bertsias, University of Crete)
Vasilis F Ntasis, Nikolaos I Panousis, Maria G Tektonidou, Emmanouil T Dermitzakis, Dimitrios T Boumpas, George K Bertsias and Christoforos Nikolaou (2020) Scientific Reports, 10(1), 16648
In a nutshell:
Systemic Lupus Erythematosus (SLE) is a complex disease with a highly variable set of symptoms, affecting a number of tissues and involving a number of cell types. Gene Expression changes in SLE have been studies from many respects but not from the point of view of genome organization. In this work we study ~200 transcriptome profiles from SLE patients and healthy individuals trying to define regions of the genome where genes are coexpressed. We detect extensive regions of gene co-expression that are interestingly changed between healthy people and SLE patients. Patients show a generally more fragmented co-expression profile which implies that disruption in gene expression patterns are generalized in the disease. More importantly, the loss of co-expression structure in disease may have significant implications in the general re-organization of gene regulation. We show that the altered co-expression structure is associated with the disruption of promoter-enhancer interactions in control of key immune and inflammatory pathways
(in collaboration with the lab of George Bertsias, University of Crete)
Vasilis F Ntasis, Nikolaos I Panousis, Maria G Tektonidou, Emmanouil T Dermitzakis, Dimitrios T Boumpas, George K Bertsias and Christoforos Nikolaou (2020) Scientific Reports, 10(1), 16648
In a nutshell:
Systemic Lupus Erythematosus (SLE) is a complex disease with a highly variable set of symptoms, affecting a number of tissues and involving a number of cell types. Gene Expression changes in SLE have been studies from many respects but not from the point of view of genome organization. In this work we study ~200 transcriptome profiles from SLE patients and healthy individuals trying to define regions of the genome where genes are coexpressed. We detect extensive regions of gene co-expression that are interestingly changed between healthy people and SLE patients. Patients show a generally more fragmented co-expression profile which implies that disruption in gene expression patterns are generalized in the disease. More importantly, the loss of co-expression structure in disease may have significant implications in the general re-organization of gene regulation. We show that the altered co-expression structure is associated with the disruption of promoter-enhancer interactions in control of key immune and inflammatory pathways
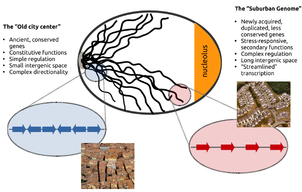
Genome Urbanization. Topological gene segregation in eukaryotes
Maria Tsochatzidou, Maria Malliarou, Joaquim Roca and Christoforos Nikolaou (2017) Genome urbanization: Clusters of topologically co-regulated genes delineate functional compartments in the genome of S. cerevisiae. Nucleic Acids Research, 45(10), 5818-5828
[Read more about this in our blog, here and here]
In a nutshell:
In all eukaryotes, the relative positions of genes are constrained by the need for complex transcriptional regulation and effective DNA replication, both of which lead to the accumulation of DNA supercoiling. In this work we performed a concise analysis of the genome architecture of S. cerevisiae, by examining the way genes respond to the inactivation of topoisomerase II. We uncovered a fundamental functional compartmentalization, according to which, conserved, essential genes are localized in gene-dense chromatin in the center of the nucleus, while stress-responsive genes tend to occupy the periphery, where broader intergenic regions allow more complex regulation. Our findings suggest a dynamic process in the evolution of genome organization that resembles segregation of modern cities into distinct neighborhoods.
Maria Tsochatzidou, Maria Malliarou, Joaquim Roca and Christoforos Nikolaou (2017) Genome urbanization: Clusters of topologically co-regulated genes delineate functional compartments in the genome of S. cerevisiae. Nucleic Acids Research, 45(10), 5818-5828
[Read more about this in our blog, here and here]
In a nutshell:
In all eukaryotes, the relative positions of genes are constrained by the need for complex transcriptional regulation and effective DNA replication, both of which lead to the accumulation of DNA supercoiling. In this work we performed a concise analysis of the genome architecture of S. cerevisiae, by examining the way genes respond to the inactivation of topoisomerase II. We uncovered a fundamental functional compartmentalization, according to which, conserved, essential genes are localized in gene-dense chromatin in the center of the nucleus, while stress-responsive genes tend to occupy the periphery, where broader intergenic regions allow more complex regulation. Our findings suggest a dynamic process in the evolution of genome organization that resembles segregation of modern cities into distinct neighborhoods.
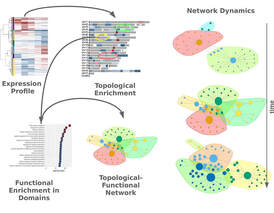
Monitoring the prolonged Tnf stimulation in space and time with topological-functional networks
Stylianos Mavropoulos Papoudas, Nikolaos Papanikolaou and Christoforos Nikolaou (2020)
In a nutshell:
A number of computational approaches exist for the analysis of gene expression data at functional level, such as GO and pathway enrichments. At the same time the positional preferences of differentially expressed genes are being overlooked and when analyzed they are studied in isolation. In this work we propose a combined approach that analyzes gene expression profiles at functional and positional levels in joint functional-topological bipartite networks. We employ this strategy in a time-dependent gene expression analysis experiment of the effect of Tnf stimulation of mouse synovial fibroblasts. Bipartite network analysis suggests that the cytokine response set by Tnf, progresses through two distinct transitions. An early generalization of the inflammatory response, that is followed by a late shutdown of immune-related functions and the redistribution of expression to developmental and cell adhesion pathways and distinct chromosomal regions. We show that the incorporation of topological information may provide additional insights in the complex propagation of Tnf activation.
Stylianos Mavropoulos Papoudas, Nikolaos Papanikolaou and Christoforos Nikolaou (2020)
In a nutshell:
A number of computational approaches exist for the analysis of gene expression data at functional level, such as GO and pathway enrichments. At the same time the positional preferences of differentially expressed genes are being overlooked and when analyzed they are studied in isolation. In this work we propose a combined approach that analyzes gene expression profiles at functional and positional levels in joint functional-topological bipartite networks. We employ this strategy in a time-dependent gene expression analysis experiment of the effect of Tnf stimulation of mouse synovial fibroblasts. Bipartite network analysis suggests that the cytokine response set by Tnf, progresses through two distinct transitions. An early generalization of the inflammatory response, that is followed by a late shutdown of immune-related functions and the redistribution of expression to developmental and cell adhesion pathways and distinct chromosomal regions. We show that the incorporation of topological information may provide additional insights in the complex propagation of Tnf activation.
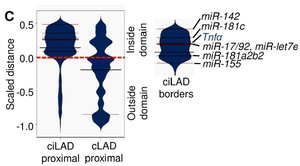
microRNAs are preferentially placed in particular nuclear positions
(in collaboration with Babis Spilianakis lab, IMBB-FORTH)
Eralda Salataj, Chrysoula Stathopoulou, Róbert A. Hafþórsson, Christoforos Nikolaou, Charalampos G. Spilianakis (2019) Developmental conservation of microRNA gene localization at the nuclear periphery. PLoS One, 14 (11): e0223759
In a nutshell:
Although there is cumulative information regarding the steady state mature microRNA levels and their respective targets, little is known about the effect of the three-dimensional chromatin architecture on the transcriptional regulation of microRNA gene loci. Here, we sought to investigate the effect of subnuclear localization on the transcriptional activation of eight murine microRNA loci in the immune system. Our results show that microRNA genes display a preferential monoallelic gene expression profile accompanied with perinuclear localization irrespectively of their transcription status or differentiation state. The expression profile and perinuclear localization are developmentally conserved while microRNA gene loci localization outside constitutive lamin associated domains is cross-species conserved. Our findings provide support for an active nuclear periphery and its role in chromatin organization of the non-coding genome.
(in collaboration with Babis Spilianakis lab, IMBB-FORTH)
Eralda Salataj, Chrysoula Stathopoulou, Róbert A. Hafþórsson, Christoforos Nikolaou, Charalampos G. Spilianakis (2019) Developmental conservation of microRNA gene localization at the nuclear periphery. PLoS One, 14 (11): e0223759
In a nutshell:
Although there is cumulative information regarding the steady state mature microRNA levels and their respective targets, little is known about the effect of the three-dimensional chromatin architecture on the transcriptional regulation of microRNA gene loci. Here, we sought to investigate the effect of subnuclear localization on the transcriptional activation of eight murine microRNA loci in the immune system. Our results show that microRNA genes display a preferential monoallelic gene expression profile accompanied with perinuclear localization irrespectively of their transcription status or differentiation state. The expression profile and perinuclear localization are developmentally conserved while microRNA gene loci localization outside constitutive lamin associated domains is cross-species conserved. Our findings provide support for an active nuclear periphery and its role in chromatin organization of the non-coding genome.

"Invisible Cities": Genome Territories with distinct structural architecture in the yeast genome.
Christoforos Nikolaou (2018) Invisible cities: segregated domains in the yeast genome with distinct structural and functional attributes. Current Genetics. 64, 247-258
[Read more about this in our blog, here]
In a nutshell:
This paper extends results from two previous works in which we had shown that yeast genes carry singular nucleosomal patterns at their promoters and that they may be divided in broader chromosomal domains with particular structural and functional characteristics. Using the above as starting point we divided the yeast genome in TAD-like domains, deduced from available public data and went on to show that they can be classified in 7 broad categories depending on the nucleosomal patterns of their promoters. These different classes of chromosomal domains are also quite different in terms of gene conservation, regulatory patterns, proximity to genomic landmarks (such as origins of DNA replication) and are unevenly distributed across chromosomes.
Christoforos Nikolaou (2018) Invisible cities: segregated domains in the yeast genome with distinct structural and functional attributes. Current Genetics. 64, 247-258
[Read more about this in our blog, here]
In a nutshell:
This paper extends results from two previous works in which we had shown that yeast genes carry singular nucleosomal patterns at their promoters and that they may be divided in broader chromosomal domains with particular structural and functional characteristics. Using the above as starting point we divided the yeast genome in TAD-like domains, deduced from available public data and went on to show that they can be classified in 7 broad categories depending on the nucleosomal patterns of their promoters. These different classes of chromosomal domains are also quite different in terms of gene conservation, regulatory patterns, proximity to genomic landmarks (such as origins of DNA replication) and are unevenly distributed across chromosomes.
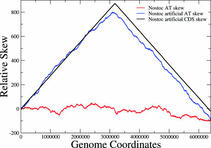
Replication mode and Genome Architecture in Prokaryote Genomes
Christoforos Nikolaou and Yannis Almirantis (2005) A Study on the correlation of nucleotide skews and the positioning of the Origin of Replication. Different modes of replication in bacterial species. Nucleic Acids Research, 33, 6816-6822
In a nutshell:
Bacterial genomes are short, circular stretches of DNA that replicate with a very well-defined process. DNA-duplication starts at a specific point of the genome and by progressing symmetrically towards both sides ends at another point that lies at a distance equal to half of the genome size. This process sort of splits the genome in two halves, each of which has very particular properties: Within either of these halves complementary nucleotides are loaded (A is more frequent than T and G more frequent than C and vice versa). Moreover, genes tend to be loaded as well in terms of transcription direction. In one half genes tend to be pointing towards one direction, while in the other they point towards the opposite. In this work we show that both these "loadings" are a direct result of evolutionary pressure imposed by the split of the genome in two parts due to the process of symmetrical DNA-replication. This work shows how a well-defined symmetrical biological process may create a sort of a conservation principle in the way suggested by Emmy Noether (see Noether's Law for more details)
Christoforos Nikolaou and Yannis Almirantis (2005) A Study on the correlation of nucleotide skews and the positioning of the Origin of Replication. Different modes of replication in bacterial species. Nucleic Acids Research, 33, 6816-6822
In a nutshell:
Bacterial genomes are short, circular stretches of DNA that replicate with a very well-defined process. DNA-duplication starts at a specific point of the genome and by progressing symmetrically towards both sides ends at another point that lies at a distance equal to half of the genome size. This process sort of splits the genome in two halves, each of which has very particular properties: Within either of these halves complementary nucleotides are loaded (A is more frequent than T and G more frequent than C and vice versa). Moreover, genes tend to be loaded as well in terms of transcription direction. In one half genes tend to be pointing towards one direction, while in the other they point towards the opposite. In this work we show that both these "loadings" are a direct result of evolutionary pressure imposed by the split of the genome in two parts due to the process of symmetrical DNA-replication. This work shows how a well-defined symmetrical biological process may create a sort of a conservation principle in the way suggested by Emmy Noether (see Noether's Law for more details)

Replication mode and compositional biases in organelle genomes
Christoforos Nikolaou and Yannis Almirantis (2006) Deviations from Chargaff's second parity rule in organellar DNA Insights into the evolution of organellar genomes. Gene , 381:34-41
In a nutshell:
On a single stranded genomic DNA molecule, complementary bases (A:T and G:C) will tend to occur with very similar frequencies as long as there are no specific asymmetries that favour one strand against the other. Such asymmetries however do exist and are related to the asymmetric processes of DNA transcription and replication. DNA replication, in particular, is extremely asymmetric in the chromosomes of eukaryotic organelles such as the mitochondria and the chloroplasts where it DNA is copied by a mechanism that replicates one of the two strands almost entirely before the other. In this work, we have analyzed the overloading of complementary bases (A vs T and G vs C), the so-called "nucleotide skews", in a large number of organelle genomes. Through this analysis we were able to show that the extent as well as the direction of the asymmetries distinguishes between the mitochondria of the three "kingdoms" (plants, animals and fungi) and chloroplasts, suggesting that the two organelles may have either originated from different organisms or adopted different replication strategies depending on their host.
Christoforos Nikolaou and Yannis Almirantis (2006) Deviations from Chargaff's second parity rule in organellar DNA Insights into the evolution of organellar genomes. Gene , 381:34-41
In a nutshell:
On a single stranded genomic DNA molecule, complementary bases (A:T and G:C) will tend to occur with very similar frequencies as long as there are no specific asymmetries that favour one strand against the other. Such asymmetries however do exist and are related to the asymmetric processes of DNA transcription and replication. DNA replication, in particular, is extremely asymmetric in the chromosomes of eukaryotic organelles such as the mitochondria and the chloroplasts where it DNA is copied by a mechanism that replicates one of the two strands almost entirely before the other. In this work, we have analyzed the overloading of complementary bases (A vs T and G vs C), the so-called "nucleotide skews", in a large number of organelle genomes. Through this analysis we were able to show that the extent as well as the direction of the asymmetries distinguishes between the mitochondria of the three "kingdoms" (plants, animals and fungi) and chloroplasts, suggesting that the two organelles may have either originated from different organisms or adopted different replication strategies depending on their host.

Linking the DNA replication machinery with nucleotide composition in bacteria
(in collaboration with Yannis Almirantis lab, NCSR "Demokritos")
Konstantinos Apostolou-Karampelis, Christoforos Nikolaou and Yannis Almirantis (2016) A novel skew analysis reveals substitution asymmetries linked to genetic code GC-biases and PolII-a subunit isoforms. DNA Research, 23(4), 353-363
In a nutshell:
In this work, based on the doctoral thesis of Konstantinos Apostolou-Karampelis we come back to the concept of DNA strand asymmetries. In a concise analysis of more than a 1000 bacterial genomes we show that tendencies in the way the two strands differ in the usage of dinucleotides may trace their evolutionary history and provide phylogenetic trees comparable to the ones obtained with standard alignment approaches. What is more important, by analyzing specific preferences within their coding regions we find a significant correlation with the way the GC-mutation pressures may be combined by the structure of the genetic code to account for the different biases observed in different genomes. Last but not least, we show that such bias can be attributed to the employment of different DNA Poll-a isoforms among bacteria.
(in collaboration with Yannis Almirantis lab, NCSR "Demokritos")
Konstantinos Apostolou-Karampelis, Christoforos Nikolaou and Yannis Almirantis (2016) A novel skew analysis reveals substitution asymmetries linked to genetic code GC-biases and PolII-a subunit isoforms. DNA Research, 23(4), 353-363
In a nutshell:
In this work, based on the doctoral thesis of Konstantinos Apostolou-Karampelis we come back to the concept of DNA strand asymmetries. In a concise analysis of more than a 1000 bacterial genomes we show that tendencies in the way the two strands differ in the usage of dinucleotides may trace their evolutionary history and provide phylogenetic trees comparable to the ones obtained with standard alignment approaches. What is more important, by analyzing specific preferences within their coding regions we find a significant correlation with the way the GC-mutation pressures may be combined by the structure of the genetic code to account for the different biases observed in different genomes. Last but not least, we show that such bias can be attributed to the employment of different DNA Poll-a isoforms among bacteria.
On nucleosome positioning and chromatin-mediated gene regulation

The Exon-Wrap. The first reported connection between chromatin structure and RNA processing
Hagen Tilgner, Christoforos Nikolaou, Sonja Althammer, Michael Sammeth, Miguel Beato, Juan Valcarcel and Roderic Guigo (2009) Nucleosome positioning as a determinant of exon recognition. Nature Structural and Molecular Biology, 16(9):996-1001.
In a nutshell:
Genomic DNA is condensed in order to fit within the cell nucleus through the formation of nucleosomes. These are protein-DNA complexes formed on DNA in positions that are partly guided by the sequence but mostly occupy the remaining space in a statistical way. In this work, we show that nucleosomes tend to be non-randomly positioned within parts of the genes that encode for proteins called exons. Not all exons are always translated into proteins and the probability of this happening depends on specific sequence signals within them. By analyzing the density of these signals and nucleosome positioning for a very large number of human exons we show that the two are inversely proportional in the sense of a compensatory effect. Remarkably, the affinity for nucleosome binding in exons is also encoded in the DNA sequence itself, which means that exons accommodate multiple layers of information apart from protein coding.
Hagen Tilgner, Christoforos Nikolaou, Sonja Althammer, Michael Sammeth, Miguel Beato, Juan Valcarcel and Roderic Guigo (2009) Nucleosome positioning as a determinant of exon recognition. Nature Structural and Molecular Biology, 16(9):996-1001.
In a nutshell:
Genomic DNA is condensed in order to fit within the cell nucleus through the formation of nucleosomes. These are protein-DNA complexes formed on DNA in positions that are partly guided by the sequence but mostly occupy the remaining space in a statistical way. In this work, we show that nucleosomes tend to be non-randomly positioned within parts of the genes that encode for proteins called exons. Not all exons are always translated into proteins and the probability of this happening depends on specific sequence signals within them. By analyzing the density of these signals and nucleosome positioning for a very large number of human exons we show that the two are inversely proportional in the sense of a compensatory effect. Remarkably, the affinity for nucleosome binding in exons is also encoded in the DNA sequence itself, which means that exons accommodate multiple layers of information apart from protein coding.
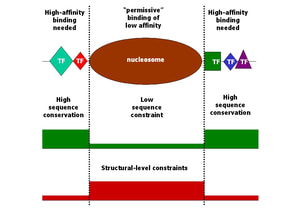
Chromatin-related sequence constraints in the genome of a simple eukaryote
Christoforos Nikolaou, Sonja Althammer, Miguel Beato and Roderic Guigo (2010) Structural constraints revealed in consistent nucleosome positions in the genome of S. cerevisiae. BMC Epigenetics and Chromatin 3:20
In a nutshell:
DNA within cells is wound around proteins called histones to form particles called nucleosomes, which in turn help the very long DNA fiber to fit within the tiny space of the cell nucleus retaining its functionality. Although nucleosomes cover more than 80% of genomic DNA, it is very difficult to predict the exact positions where they will bind on the linear sequence, an issue of great importance given that they compete in this with other important DNA-binding proteins. In this study we show, by comparing two independently performed nucleosome localization studies in yeast, that only one out of ten nucleosomes actually binds to a specific stretch of DNA with the rest occupying the remaining space in a more or less statistical manner. We further show that for this 10% of well-positioned nucleosomes the DNA "guidance" actually comes from the part of the DNA that remains unbound by nucleosomes and acts as "exclusion-sequences" forcing the nucleosome to bind in between them.
Christoforos Nikolaou, Sonja Althammer, Miguel Beato and Roderic Guigo (2010) Structural constraints revealed in consistent nucleosome positions in the genome of S. cerevisiae. BMC Epigenetics and Chromatin 3:20
In a nutshell:
DNA within cells is wound around proteins called histones to form particles called nucleosomes, which in turn help the very long DNA fiber to fit within the tiny space of the cell nucleus retaining its functionality. Although nucleosomes cover more than 80% of genomic DNA, it is very difficult to predict the exact positions where they will bind on the linear sequence, an issue of great importance given that they compete in this with other important DNA-binding proteins. In this study we show, by comparing two independently performed nucleosome localization studies in yeast, that only one out of ten nucleosomes actually binds to a specific stretch of DNA with the rest occupying the remaining space in a more or less statistical manner. We further show that for this 10% of well-positioned nucleosomes the DNA "guidance" actually comes from the part of the DNA that remains unbound by nucleosomes and acts as "exclusion-sequences" forcing the nucleosome to bind in between them.
Nucleosomes and Topoisomerases. Distinct promoter architectures in topoII-regulated genes
(in collaboration with the lab of Joaquim Roca, IBMB-CSIC, Barcelona)
Christoforos Nikolaou, Ignacio Bermudez Herrojo, Chaysavanah, Garcia-Martinez, Roderic Guigo, Perez-Ortin J.E. and Joaquim Roca (2013) Topoisomerase II regulates yeast genes with singular promoter architectures. Nucleic Acids Research, 41 (20) 9243-9256
In a nutshell:
Cellular processes taking place in the chromatin component of the nucleus, impose various distortions in the "thread" of the DNA molecule. Such knots and entanglements are resolved in the cell by specific molecules, called topoisomerases, the absence of which has great impact on the genome's function. In this work we show that when topoisomerases are absent, not all genes are affected in the same way. In fact some genes are "switched on" and others are "switched off" and this largely depends on local properties of the sequence and the structure of the promoter, that is the region of the DNA close to the gene that affects its regulation. We further show that, depending on their chromatin structure, these genes also have different functions in the cell and attract different kinds of DNA-binding proteins.
(in collaboration with the lab of Joaquim Roca, IBMB-CSIC, Barcelona)
Christoforos Nikolaou, Ignacio Bermudez Herrojo, Chaysavanah, Garcia-Martinez, Roderic Guigo, Perez-Ortin J.E. and Joaquim Roca (2013) Topoisomerase II regulates yeast genes with singular promoter architectures. Nucleic Acids Research, 41 (20) 9243-9256
In a nutshell:
Cellular processes taking place in the chromatin component of the nucleus, impose various distortions in the "thread" of the DNA molecule. Such knots and entanglements are resolved in the cell by specific molecules, called topoisomerases, the absence of which has great impact on the genome's function. In this work we show that when topoisomerases are absent, not all genes are affected in the same way. In fact some genes are "switched on" and others are "switched off" and this largely depends on local properties of the sequence and the structure of the promoter, that is the region of the DNA close to the gene that affects its regulation. We further show that, depending on their chromatin structure, these genes also have different functions in the cell and attract different kinds of DNA-binding proteins.
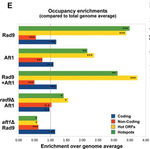
A DNA-damage checkpoint protein is "safeguarding" fragile genomic positions even in the absence of DNA damage
(in collaboration with the lab of Despina Alexandraki, IMBB-FORTH)
Christos Andreadis, Christoforos Nikolaou, George Fragiadakis, Georgia Tsiliki and Despina Alexandraki (2014) Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Research, 42(20), 12650-12667
In a nutshell:
Genomic DNA undergoes a number of processes that render it susceptible to single and double-stranded breaks. Such "DNA damage" as it is called may have severe consequences for the fate of the cells and so organisms have developed sophisticated mechanisms for "sounding the alarm" in case DNA damage occurs. In this work, a collaboration with the Laboratory of Gene Expression at IMBB-FORTH, we show that such mechanisms are even more sophisticated than simply signalling for damage. The paper, part of the PhD thesis of Christos Andreadis shows that a yeast DNA-damage checkpoint protein, Rad9, is preferentially "stationed" at regions that are expected to undergo increased stress in terms of DNA damage, such as genes that are transcribed more often or regions where DNA "opens" for replication or recombination. In this sense, this work points out to a model of DNA surveillance, under which DNA-damage checkpoint proteins assume a more "preventive" role than initially thought.
(in collaboration with the lab of Despina Alexandraki, IMBB-FORTH)
Christos Andreadis, Christoforos Nikolaou, George Fragiadakis, Georgia Tsiliki and Despina Alexandraki (2014) Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Research, 42(20), 12650-12667
In a nutshell:
Genomic DNA undergoes a number of processes that render it susceptible to single and double-stranded breaks. Such "DNA damage" as it is called may have severe consequences for the fate of the cells and so organisms have developed sophisticated mechanisms for "sounding the alarm" in case DNA damage occurs. In this work, a collaboration with the Laboratory of Gene Expression at IMBB-FORTH, we show that such mechanisms are even more sophisticated than simply signalling for damage. The paper, part of the PhD thesis of Christos Andreadis shows that a yeast DNA-damage checkpoint protein, Rad9, is preferentially "stationed" at regions that are expected to undergo increased stress in terms of DNA damage, such as genes that are transcribed more often or regions where DNA "opens" for replication or recombination. In this sense, this work points out to a model of DNA surveillance, under which DNA-damage checkpoint proteins assume a more "preventive" role than initially thought.
On complex diseases, transcription and epigenetics
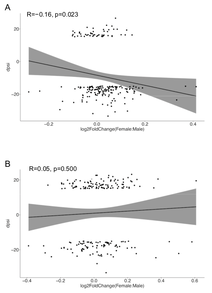
Extensive Alternative Splicing Patterns in Systemic Lupus Erythematosus Highlight Sexual Differences
(in collaboration with the lab of George Bertsias at the Medical School, University of Crete)
Despoina Kosmara, Sofia Papanikolaou, Christoforos Nikolaou and George Bertsias (2023) Cells, 12, 2678
In a nutshell:
Substantial evidence highlights divergences in immune responses between men and women. Women are more susceptible to autoimmunity, whereas men suffer from the more severe presentation of autoimmune disorders. The molecular mechanism of this sexual dimorphism remains elusive. Herein, we conducted a comprehensive analysis of sex differences in whole-blood gene expression focusing on alternative splicing (AS) events in systemic lupus erythematosus (SLE), which is a prototype sex-biased disease. This study included 79 SLE patients with active disease and 58 matched
healthy controls who underwent whole-blood RNA sequencing. Sex differences in splicing events were widespread, existent in both SLE and a healthy state. However, we observed distinct gene sets and molecular pathways targeted by sex-dependent AS in SLE patients as compared to healthy subjects, as well as a notable sex dissimilarity in intron retention events. Sexually differential spliced genes specific to SLE patients were enriched for dynamic cellular processes including chromatin remodeling, stress and inflammatory responses. Remarkably, the extent of sexual differences in AS in the SLE patients and healthy individuals exceeded those in gene expression. Overall, this study reveals an unprecedent variation in sex-dependent splicing events in SLE and the healthy state, with potential implications for understanding the molecular basis of sexual dimorphism in autoimmunity.
(in collaboration with the lab of George Bertsias at the Medical School, University of Crete)
Despoina Kosmara, Sofia Papanikolaou, Christoforos Nikolaou and George Bertsias (2023) Cells, 12, 2678
In a nutshell:
Substantial evidence highlights divergences in immune responses between men and women. Women are more susceptible to autoimmunity, whereas men suffer from the more severe presentation of autoimmune disorders. The molecular mechanism of this sexual dimorphism remains elusive. Herein, we conducted a comprehensive analysis of sex differences in whole-blood gene expression focusing on alternative splicing (AS) events in systemic lupus erythematosus (SLE), which is a prototype sex-biased disease. This study included 79 SLE patients with active disease and 58 matched
healthy controls who underwent whole-blood RNA sequencing. Sex differences in splicing events were widespread, existent in both SLE and a healthy state. However, we observed distinct gene sets and molecular pathways targeted by sex-dependent AS in SLE patients as compared to healthy subjects, as well as a notable sex dissimilarity in intron retention events. Sexually differential spliced genes specific to SLE patients were enriched for dynamic cellular processes including chromatin remodeling, stress and inflammatory responses. Remarkably, the extent of sexual differences in AS in the SLE patients and healthy individuals exceeded those in gene expression. Overall, this study reveals an unprecedent variation in sex-dependent splicing events in SLE and the healthy state, with potential implications for understanding the molecular basis of sexual dimorphism in autoimmunity.
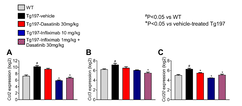
Targeting senescence and inflammation in chronic destructive TNF-driven joint pathology
(in collaboration with the lab of Petros Sfikakis, Medical School, University of Athens)
Nikolaos I. Vlachogiannis, Konstantinos Evangelou, Lydia Ntari, Christoforos Nikolaou, Maria C. Denis, Niki Karagianni, Dimitris Veroutis, Vassilis Gorgoulis, George Kollias, Petros P. Sfikakis. (2023) Mechanisms of Ageing and Development 214, 111856
In a nutshell:
Following from a previous work (Ntari et al, 2021) in which we had shown that administration of the senolytic Dasatinib abolishes arthritis in the human TNF transgenic mouse model of chronic destructive arthritis when given in combination with a sub-therapeutic dose of the anti-TNF mAb Infliximab (1 mg/kg), we here found that while the number of senescent chondrocytes (GL13+/Ki67-), assessed according to guideline algorithmic approaches, was not affected by either Dasatinib or sub-therapeutic Infliximab monotherapies, their combination reduced senescent chondrocytes by 50 %, which was comparable to levels observed with therapeutic Infliximab monotherapy (10 mg/kg). This combination therapy also reduced the expression of multiple factors of senescence-associated secretory phenotype in arthritic joints. Studies to elucidate the interplay of inflammation and senescence may help in optimizing treatment strategies also for age-related pathologies characterized by chronic low-grade joint inflammation.
(in collaboration with the lab of Petros Sfikakis, Medical School, University of Athens)
Nikolaos I. Vlachogiannis, Konstantinos Evangelou, Lydia Ntari, Christoforos Nikolaou, Maria C. Denis, Niki Karagianni, Dimitris Veroutis, Vassilis Gorgoulis, George Kollias, Petros P. Sfikakis. (2023) Mechanisms of Ageing and Development 214, 111856
In a nutshell:
Following from a previous work (Ntari et al, 2021) in which we had shown that administration of the senolytic Dasatinib abolishes arthritis in the human TNF transgenic mouse model of chronic destructive arthritis when given in combination with a sub-therapeutic dose of the anti-TNF mAb Infliximab (1 mg/kg), we here found that while the number of senescent chondrocytes (GL13+/Ki67-), assessed according to guideline algorithmic approaches, was not affected by either Dasatinib or sub-therapeutic Infliximab monotherapies, their combination reduced senescent chondrocytes by 50 %, which was comparable to levels observed with therapeutic Infliximab monotherapy (10 mg/kg). This combination therapy also reduced the expression of multiple factors of senescence-associated secretory phenotype in arthritic joints. Studies to elucidate the interplay of inflammation and senescence may help in optimizing treatment strategies also for age-related pathologies characterized by chronic low-grade joint inflammation.

Extensive Changes in Transcription Dynamics Reflected on Alternative Splicing Events in Systemic Lupus Erythematosus Patients
Sofia Papanikolaou, George K. Bertsias and Christoforos Nikolaou*. (2021) Genes, 12(8), 1260
In a nutshell:
In addition to increasing the complexity of the transcriptional output, alternative RNA splicing can lead to the reduction of mRNA translation or the production of non-functional or malfunctional proteins, thus representing a vital component of the gene regulation process. Herein, we set out to detect and characterize alternative splicing events that occur in whole-blood samples of patients with Systemic Lupus Erythematosus (SLE) as compared to healthy counterparts. Through the implementation of a computational pipeline on published RNA-sequencing data, we identified extensive changes in the transcription dynamics affecting a large number of genes. We found a predominance of intron retention events, with the majority introducing premature stop codons, suggestive of gene repression, in both inactive and active SLE patient samples. Alternative splicing affected a distinct set of genes from the ones detected as differentially expressed in the same comparisons, while alternatively spliced genes tended to reside in genome areas associated with increased gene co-expression. Functional analysis of genes affected by alternative splicing pointed towards particular functions related to metabolism and histone acetylation as of potential interest. Together, our findings underline the importance of incorporating alternative splicing analyses in the context of molecular characterization of complex diseases such as SLE.
Sofia Papanikolaou, George K. Bertsias and Christoforos Nikolaou*. (2021) Genes, 12(8), 1260
In a nutshell:
In addition to increasing the complexity of the transcriptional output, alternative RNA splicing can lead to the reduction of mRNA translation or the production of non-functional or malfunctional proteins, thus representing a vital component of the gene regulation process. Herein, we set out to detect and characterize alternative splicing events that occur in whole-blood samples of patients with Systemic Lupus Erythematosus (SLE) as compared to healthy counterparts. Through the implementation of a computational pipeline on published RNA-sequencing data, we identified extensive changes in the transcription dynamics affecting a large number of genes. We found a predominance of intron retention events, with the majority introducing premature stop codons, suggestive of gene repression, in both inactive and active SLE patient samples. Alternative splicing affected a distinct set of genes from the ones detected as differentially expressed in the same comparisons, while alternatively spliced genes tended to reside in genome areas associated with increased gene co-expression. Functional analysis of genes affected by alternative splicing pointed towards particular functions related to metabolism and histone acetylation as of potential interest. Together, our findings underline the importance of incorporating alternative splicing analyses in the context of molecular characterization of complex diseases such as SLE.
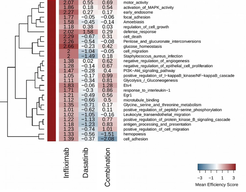
Combination of subtherapeutic anti-TNF dose with dasatinib restores clinical and molecular arthritogenic profiles better than standard anti-TNF treatment
(in collaboration with Biomedcode SA)
Lydia Ntari, Christoforos Nikolaou, Ksanthi Kranidioti, Dimitra Papadopoulou, Eleni Christodoulou-Vafeiadou, Panagiotis Chouvardas, Florian Meier, Christina Geka, Maria C. Denis, Niki Karagianni & George Kollias. Journal of Translational Medicine, (2021), 19, 165
In a nutshell:
New medications for Rheumatoid Arthritis (RA) have emerged in the last decades, including Disease Modifying Antirheumatic Drugs (DMARDs) and biologics. However, there is no known cure, since a significant proportion of patients remain or become non-responders to current therapies. The development of new mode-of-action treatment schemes involving combination therapies could prove successful for the treatment of a greater number of RA patients. We investigated the effect of the Tyrosine Kinase inhibitors (TKIs) dasatinib and bosutinib, on the human TNF-dependent Tg197 arthritis mouse model. The inhibitors were administered either as a monotherapy or in combination with a subtherapeutic dose of anti-hTNF biologics and their therapeutic effect was assessed clinically, histopathologically as well as via gene expression analysis and was compared to that of an efficient TNF monotherapy. Combination of dasatinib with a subtherapeutic dose of anti-hTNF biologic agents, resulted in a synergistic inhibitory effect abolishing all arthritis symptoms. Gene expression analysis of whole joint tissue of Tg197 mice revealed that the combination of dasatinib with a low subtherapeutic dose of Infliximab most efficiently restores the pathogenic gene expression profile to that of the healthy state compared to either treatment administered as a monotherapy. Our findings show that dasatinib exhibits a therapeutic effect in TNF-driven arthritis and can act in synergy with a subtherapeutic anti-hTNF dose to effectively treat the clinical and histopathological signs of the pathology. Potential clinical applications of combination therapies with kinase inhibitors and anti-TNF agents may provide an interesting alternative to high-dose anti-hTNF monotherapy and increase the number of patients responding to treatment.
(in collaboration with Biomedcode SA)
Lydia Ntari, Christoforos Nikolaou, Ksanthi Kranidioti, Dimitra Papadopoulou, Eleni Christodoulou-Vafeiadou, Panagiotis Chouvardas, Florian Meier, Christina Geka, Maria C. Denis, Niki Karagianni & George Kollias. Journal of Translational Medicine, (2021), 19, 165
In a nutshell:
New medications for Rheumatoid Arthritis (RA) have emerged in the last decades, including Disease Modifying Antirheumatic Drugs (DMARDs) and biologics. However, there is no known cure, since a significant proportion of patients remain or become non-responders to current therapies. The development of new mode-of-action treatment schemes involving combination therapies could prove successful for the treatment of a greater number of RA patients. We investigated the effect of the Tyrosine Kinase inhibitors (TKIs) dasatinib and bosutinib, on the human TNF-dependent Tg197 arthritis mouse model. The inhibitors were administered either as a monotherapy or in combination with a subtherapeutic dose of anti-hTNF biologics and their therapeutic effect was assessed clinically, histopathologically as well as via gene expression analysis and was compared to that of an efficient TNF monotherapy. Combination of dasatinib with a subtherapeutic dose of anti-hTNF biologic agents, resulted in a synergistic inhibitory effect abolishing all arthritis symptoms. Gene expression analysis of whole joint tissue of Tg197 mice revealed that the combination of dasatinib with a low subtherapeutic dose of Infliximab most efficiently restores the pathogenic gene expression profile to that of the healthy state compared to either treatment administered as a monotherapy. Our findings show that dasatinib exhibits a therapeutic effect in TNF-driven arthritis and can act in synergy with a subtherapeutic anti-hTNF dose to effectively treat the clinical and histopathological signs of the pathology. Potential clinical applications of combination therapies with kinase inhibitors and anti-TNF agents may provide an interesting alternative to high-dose anti-hTNF monotherapy and increase the number of patients responding to treatment.
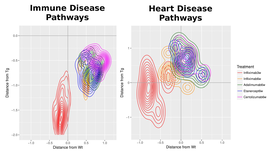
Dissecting gene expression profiles of pharmacological intervention in Rheumatoid Arthritis
(in collaboration with Biomedcode SA)
Niki Karagianni et al, including Christoforos Nikolaou (2019) An integrative transcriptome analysis framework for drug efficacy and similarity reveals drug-specific signatures of anti-TNF treatment in a mouse model of inflammatory polyarthritis. PLoS Computational Biology 15(5): e1006933.
In a nutshell:
e employed an extensive gene expression profiling of anti-TNF agents in the mice suffering from inflammatory polyarthritis, closely resembling human rheumatoid arthritis. By analyzing large numbers of biological replicates we found significant differences between four tested anti-TNF substances at both qualitative and quantitative levels. Through the combination of multi-level functional enrichment analysis and the implementation of Random Forest classification models we show that a subset of the tested substances are more prone to modify functions related to up-regulated genes associated with inflammation and the immune response. A different drug subset, on the other hand, is more effective in restoring normal gene expression levels for down-regulated pathways associated with rheumatoid arthritis commorbidities such as cardiomyopathies. Together, our results point towards a molecular basis for the observed variability of response to the treatment of Rheumatoid Arthritis.
(in collaboration with Biomedcode SA)
Niki Karagianni et al, including Christoforos Nikolaou (2019) An integrative transcriptome analysis framework for drug efficacy and similarity reveals drug-specific signatures of anti-TNF treatment in a mouse model of inflammatory polyarthritis. PLoS Computational Biology 15(5): e1006933.
In a nutshell:
e employed an extensive gene expression profiling of anti-TNF agents in the mice suffering from inflammatory polyarthritis, closely resembling human rheumatoid arthritis. By analyzing large numbers of biological replicates we found significant differences between four tested anti-TNF substances at both qualitative and quantitative levels. Through the combination of multi-level functional enrichment analysis and the implementation of Random Forest classification models we show that a subset of the tested substances are more prone to modify functions related to up-regulated genes associated with inflammation and the immune response. A different drug subset, on the other hand, is more effective in restoring normal gene expression levels for down-regulated pathways associated with rheumatoid arthritis commorbidities such as cardiomyopathies. Together, our results point towards a molecular basis for the observed variability of response to the treatment of Rheumatoid Arthritis.

Intestinal myofibroblast-specific Tpl2-Cox-2-PGE 2 pathway links innate sensing to epithelial homeostasis.
(in collaboration with George Kollias lab, BSRC "Alexander Flemming")
Manolis Roulis, Christoforos Nikolaou et al. (2015) PNAS, 111, 4658-4667
In a nutshell:
Tumor progression locus-2 (Tpl2) is a proinflammatory gene genetically associated with inflammatory bowel diseases. This study provides a mechanistic interpretation for this association showing a dominant Tpl2-mediated homeostatic mechanism protecting mice from epithelial injury-induced colitis. CG2 has contributed in this work through the implementation of novel tools in the analysis of RNA Sequencing data that led to the identification of Tpl2-interacting proteins that pave the way for the elucidation of its homeostatic function.
(in collaboration with George Kollias lab, BSRC "Alexander Flemming")
Manolis Roulis, Christoforos Nikolaou et al. (2015) PNAS, 111, 4658-4667
In a nutshell:
Tumor progression locus-2 (Tpl2) is a proinflammatory gene genetically associated with inflammatory bowel diseases. This study provides a mechanistic interpretation for this association showing a dominant Tpl2-mediated homeostatic mechanism protecting mice from epithelial injury-induced colitis. CG2 has contributed in this work through the implementation of novel tools in the analysis of RNA Sequencing data that led to the identification of Tpl2-interacting proteins that pave the way for the elucidation of its homeostatic function.
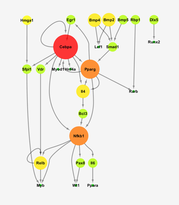
Inferring Regulatory Networks from Gene Expression Profiles
(in collaboration with George Kollias lab, BSRC "Alexander Flemming")
Panagiotis Chouvardas, George Kollias and Christoforos Nikolaou (2016) Inferring active regulatory networks from gene expression data using a combination of prior knowledge and enrichment analysis. BMC Bioinformatics, 17: 181
In a nutshell:
Even though we can accurately measure and assess the expression levels of the complete set of an organism's genes, we are still far from being able to say how these genes regulated each other. In this work, we use prior knowledge of gene-gene interactions to compile two large reference networks (human and mouse) and a simple hierarchical inference model that uses gene expression values to tell us which gene activates which. As a result we present a bioinformatics tool that takes a) a reference network and b) a gene expression profile and provides a regulatory subnetwork that would best describe the observed expression values. This tool (RNEA) can be used to provide valuable insight on the regulatory hierarchy given a gene expression experiment.
(in collaboration with George Kollias lab, BSRC "Alexander Flemming")
Panagiotis Chouvardas, George Kollias and Christoforos Nikolaou (2016) Inferring active regulatory networks from gene expression data using a combination of prior knowledge and enrichment analysis. BMC Bioinformatics, 17: 181
In a nutshell:
Even though we can accurately measure and assess the expression levels of the complete set of an organism's genes, we are still far from being able to say how these genes regulated each other. In this work, we use prior knowledge of gene-gene interactions to compile two large reference networks (human and mouse) and a simple hierarchical inference model that uses gene expression values to tell us which gene activates which. As a result we present a bioinformatics tool that takes a) a reference network and b) a gene expression profile and provides a regulatory subnetwork that would best describe the observed expression values. This tool (RNEA) can be used to provide valuable insight on the regulatory hierarchy given a gene expression experiment.
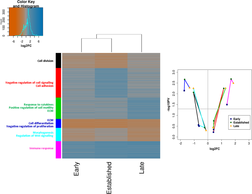
The Rheumatome: A multi-level genomic analysis of synovial fibroblasts from a TNF-transgenic mouse model of Rheumatoid Arthritis
(in collaboration with George Kollias lab, BSRC "Alexander Flemming")
Evangelos Ntougkos, Panagiotis Chouvardas, Fani Roumelioti, et al., including Christoforos Nikolaou and George Kollias (2017), Arthritis and Rheumatology, 69(8), 1588-1600
By studying the gene expression, DNA methylation and activating epigenetic patterns in synovial fibroblasts of a TNF-transgenic mouse and in three time points corresponding to three distinct disease stages we provide a concsise overview of gene clusters, functions and biological pathways that are modulated as disease progresses both at gene expression and at epigenetic levels. Comparison with the human condition (RA patient samples) shows extensive alignments which suggests that ours may serve as a valuable dataset for the study of RA. Besides its function as a data repository, our work reveals a number of interesting aspects of disease progression including functions that are differentially modulated across time points as well as the corroboration of a number of gene-drivers of the disease such as PPARγ, WNT16 and SP7.
(in collaboration with George Kollias lab, BSRC "Alexander Flemming")
Evangelos Ntougkos, Panagiotis Chouvardas, Fani Roumelioti, et al., including Christoforos Nikolaou and George Kollias (2017), Arthritis and Rheumatology, 69(8), 1588-1600
By studying the gene expression, DNA methylation and activating epigenetic patterns in synovial fibroblasts of a TNF-transgenic mouse and in three time points corresponding to three distinct disease stages we provide a concsise overview of gene clusters, functions and biological pathways that are modulated as disease progresses both at gene expression and at epigenetic levels. Comparison with the human condition (RA patient samples) shows extensive alignments which suggests that ours may serve as a valuable dataset for the study of RA. Besides its function as a data repository, our work reveals a number of interesting aspects of disease progression including functions that are differentially modulated across time points as well as the corroboration of a number of gene-drivers of the disease such as PPARγ, WNT16 and SP7.
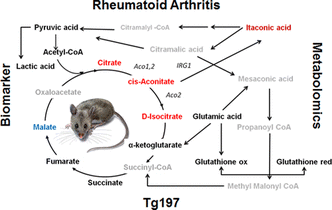
Targeted Metabolic Profiling of the Tg197 Mouse Model Reveals Itaconic Acid as a Marker of Rheumatoid Arthritis.
(in collaboration with George Kollias lab, BSRC "Alexander Flemming" and Biomedcode SA)
Filippos Michopoulos, Niki Karagianni, Nichola M. Whalley, Mike A. Firth, Christoforos Nikolaou, Ian D. Wilson, Susan E. Critchlow, George Kollias, and Georgios A. Theodoridis, (2016), Journal of Proteome Research, 15(12), 4579-4590.
In a nutshell:
Rheumatoid arthritis is a progressive, highly debilitating disease where early diagnosis, enabling rapid clinical intervention, would provide obvious benefits to patients, healthcare systems, and society. In this work we combined a targeted metabolic profiling method and gene expression data in the Tg197 arthritis mouse model, in order to define possible metabolite biomarkers. Our analysis highlighted itaconic acid, a metabolite which can be produced in a one-step reaction from cis-aconitate (a member of the Krebs cycle) as a potential marker of the disease. Itaconate levels were significantly higher in serum and urine samples of diseased mice, while they dropped considerably upon treatment of the animals with a TNF antibody (infliximab). Besides their potential practical importance, our results highlight the relevance of the citric acid cycle in disease progression. Indeed, as shown through gene expression analyses, various enzymes of the Krebs cycle where found to be dowregulated in the diseased state, which points to a general suppression of this metabolic path in RA, a consequence of which may be the accumulation of cycle's intermediates such as isocitrate and cis-aconitate, which then give rise to itaconic acid.
(in collaboration with George Kollias lab, BSRC "Alexander Flemming" and Biomedcode SA)
Filippos Michopoulos, Niki Karagianni, Nichola M. Whalley, Mike A. Firth, Christoforos Nikolaou, Ian D. Wilson, Susan E. Critchlow, George Kollias, and Georgios A. Theodoridis, (2016), Journal of Proteome Research, 15(12), 4579-4590.
In a nutshell:
Rheumatoid arthritis is a progressive, highly debilitating disease where early diagnosis, enabling rapid clinical intervention, would provide obvious benefits to patients, healthcare systems, and society. In this work we combined a targeted metabolic profiling method and gene expression data in the Tg197 arthritis mouse model, in order to define possible metabolite biomarkers. Our analysis highlighted itaconic acid, a metabolite which can be produced in a one-step reaction from cis-aconitate (a member of the Krebs cycle) as a potential marker of the disease. Itaconate levels were significantly higher in serum and urine samples of diseased mice, while they dropped considerably upon treatment of the animals with a TNF antibody (infliximab). Besides their potential practical importance, our results highlight the relevance of the citric acid cycle in disease progression. Indeed, as shown through gene expression analyses, various enzymes of the Krebs cycle where found to be dowregulated in the diseased state, which points to a general suppression of this metabolic path in RA, a consequence of which may be the accumulation of cycle's intermediates such as isocitrate and cis-aconitate, which then give rise to itaconic acid.
On gene regulation in differentiation and cancer
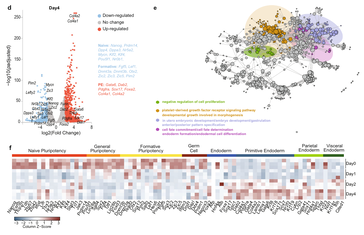
The Wnt/TCF7L1 transcriptional repressor axis drives primitive endoderm formation by antagonizing naive and formative pluripotency.
(in collaboration with the lab of Frederic Lluis Vinas at KU Leuven)
Paraskevi Athanasouli, Martina Balli, Anchel De Jaime-Soguero, Annekatrien Boel, Sofia Papanikolaou, Bernard K. van der Veer, Adrian Janiszewski, Tijs Vanhessche, Annick Francis, Youssef El Laithy, Antonio Lo Nigro, Francesco Aulicino, Kian Peng Koh, Vincent Pasque, Maria Pia Cosma, Catherine Verfaillie, An Zwijsen, Björn Heindryckx, Christoforos Nikolaou and Frederic Lluis. (2023). Nature Communications, 14: 1210.
In a nutshell:
Early during preimplantation development and in heterogeneous mouse embryonic stem cells (mESC) culture, pluripotent cells are specified towards either the primed epiblast or the primitive endoderm (PE) lineage. Canonical Wnt signaling is crucial for safeguarding naive pluripotency and embryo implantation, yet the role and relevance of canonical Wnt inhibition during early mammalian development remains unknown. Here, we demonstrate that transcriptional repression exerted by Wnt/TCF7L1 promotes PE differentiation of mESCs and in preimplantation inner cell mass. Time-series RNA sequencing and promoter occupancy data reveal that TCF7L1 binds and represses genes encoding essential naive pluripotency factors and indispensable regulators of the formative pluripotency program, including Otx2 and Lef1. Consequently, TCF7L1 promotes pluripotency exit and suppresses epiblast lineage formation, thereby driving cells into PE specification. Conversely, TCF7L1 is required for PE specification as deletion of Tcf7l1 abrogates PE differentiation without restraining epiblast priming. Taken together, our study underscores the importance of transcriptional Wnt inhibition in regulating lineage specification in ESCs and preimplantation embryo development as well as identifies TCF7L1 as key regulator of this process.
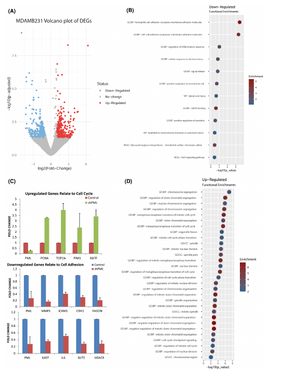
Promyelocytic leukemia protein regulates angiogenesis and epithelial–mesenchymal transition to limit metastasis in MDA-MB-231 breast cancer cells
(in collaboration with the labs of Niki Kretsovali and Joseph Papamatheakis at IMBB-FORTH)
Amalia P. Vogiatzoglou, Syrago Spanou, Nikoleta Sachini, Elias Drakos, Christoforos Nikolaou, Takis Makatounakis, Androniki Kretsovali, Joseph Papamatheakis (2023). Molecular Oncology, 17:10
In a nutshell:
Promyelocytic leukemia protein (PML) modulates diverse cell functions that contribute to both tumor suppressor and pro-oncogenic effects, depending on the cellular context. In this work, we show that PML knockdown (KD) in MDA-MB-231, but not MCF7, breast cancer cells, prolonged stem-cell-like survival, and increased cell proliferation and migration, which is in line with gene-enrichment results from their RNA sequencing analysis. Increased migration was accompanied by higher levels of the epithelial–mesenchymal transition (EMT) regulator Twist-related protein 2 (TWIST2). We showed here that PML binds to TWIST2 via its basic helix–loop–helix (bHLH) region and functionally interferes with the suppression of the epithelial target of TWIST2, CD24. In addition, PML ablation in MDA-MB-231 cells led to higher protein levels of hypoxia-inducible factor 1-alpha (HIF1a), resulting in a higher cell hypoxic response. Functionally, PML directly suppressed the induction of the HIF1a target gene vascular endothelial growth factor A (VEGFa).Collectively, we show that PML suppresses the cancer aggressive behavior by multiple mechanisms that impede both the HIF–hypoxia–angiogenic and EMT pathways.
(in collaboration with the labs of Niki Kretsovali and Joseph Papamatheakis at IMBB-FORTH)
Amalia P. Vogiatzoglou, Syrago Spanou, Nikoleta Sachini, Elias Drakos, Christoforos Nikolaou, Takis Makatounakis, Androniki Kretsovali, Joseph Papamatheakis (2023). Molecular Oncology, 17:10
In a nutshell:
Promyelocytic leukemia protein (PML) modulates diverse cell functions that contribute to both tumor suppressor and pro-oncogenic effects, depending on the cellular context. In this work, we show that PML knockdown (KD) in MDA-MB-231, but not MCF7, breast cancer cells, prolonged stem-cell-like survival, and increased cell proliferation and migration, which is in line with gene-enrichment results from their RNA sequencing analysis. Increased migration was accompanied by higher levels of the epithelial–mesenchymal transition (EMT) regulator Twist-related protein 2 (TWIST2). We showed here that PML binds to TWIST2 via its basic helix–loop–helix (bHLH) region and functionally interferes with the suppression of the epithelial target of TWIST2, CD24. In addition, PML ablation in MDA-MB-231 cells led to higher protein levels of hypoxia-inducible factor 1-alpha (HIF1a), resulting in a higher cell hypoxic response. Functionally, PML directly suppressed the induction of the HIF1a target gene vascular endothelial growth factor A (VEGFa).Collectively, we show that PML suppresses the cancer aggressive behavior by multiple mechanisms that impede both the HIF–hypoxia–angiogenic and EMT pathways.
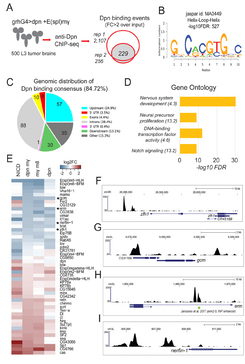
Dissecting Hes-centered transcriptional networks in neural stem cell maintenance and tumorigenesis in Drosophila
(in collaboration with Christos Delidakis' lab, at IMBB-FORTH)
Srivathsa S Magadi, Chrysanthi Voutyraki, Gerasimos Anagnostopoulos, Evanthia Zacharioudaki, Ioanna K Poutakidou, Christina Efraimoglou, Margarita Stapountzi, Vasiliki Theodorou, Christoforos Nikolaou, Konstantinos A Koumbanakis, John F Fullard and Christos Delidakis (2020), Development, 147(22):dev191544
In a nutshell:
Neural stem cells divide during embryogenesis and juvenile life to generate the entire complement of neurons and glia in the nervous system of vertebrates and invertebrates. Here, we show that excessive activation of Notch or overexpression of its direct targets of the Hes family causes stem-cell hyperplasias in the Drosophila larval central nervous system, which can progress to malignant tumours after allografting to adult hosts. We combined transcriptomic data from these hyperplasias with chromatin occupancy data for Dpn, a Hes transcription factor, to identify genes regulated by Hes factors in this process. We show that the Notch/Hes axis represses a cohort of transcription factor genes. We describe the impact of two of these 'anti-stemness' factors, Zfh1 and Gcm, on Notch/Hes-triggered tumorigenesis.
(in collaboration with Christos Delidakis' lab, at IMBB-FORTH)
Srivathsa S Magadi, Chrysanthi Voutyraki, Gerasimos Anagnostopoulos, Evanthia Zacharioudaki, Ioanna K Poutakidou, Christina Efraimoglou, Margarita Stapountzi, Vasiliki Theodorou, Christoforos Nikolaou, Konstantinos A Koumbanakis, John F Fullard and Christos Delidakis (2020), Development, 147(22):dev191544
In a nutshell:
Neural stem cells divide during embryogenesis and juvenile life to generate the entire complement of neurons and glia in the nervous system of vertebrates and invertebrates. Here, we show that excessive activation of Notch or overexpression of its direct targets of the Hes family causes stem-cell hyperplasias in the Drosophila larval central nervous system, which can progress to malignant tumours after allografting to adult hosts. We combined transcriptomic data from these hyperplasias with chromatin occupancy data for Dpn, a Hes transcription factor, to identify genes regulated by Hes factors in this process. We show that the Notch/Hes axis represses a cohort of transcription factor genes. We describe the impact of two of these 'anti-stemness' factors, Zfh1 and Gcm, on Notch/Hes-triggered tumorigenesis.

A role for mouse Promyelocytic Leukemia Protein in Cell Differentiation
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Christiana Hadjimichael, Konstantina Chanoumidou, Christoforos Nikolaou, Antonios Klonizakis, Gesthimani-Ioanna Theodosi, Takis Makatounakis, Joseph Papamatheakis, Androniki Kretsovali (2017), Stem Cell Reports, 8(5), 1366-1378
In a nutshell:
Promyelocytic leukemia protein (PML) is a tumor suppressor protein and the major constituent of PML-nuclear bodies, which are known to have a number of regulatory cellular functions, including involvement in programmed cell death, genome stability, antiviral effects and controlling cell division. In this work, we show that PML also plays a major role in the maintenance of pluripotency in mouse ES cells, exhibiting its control on a number of pathways including that of TGF-β at an early stage. The main point of this work is that it provides a link between a protein that has been extensively studied in a different context (its role in cell cycle and apoptosis) and a developmental process such as cell differentiation. Our group's involvement in this work was principally related to the analysis of gene expression data, the identification and prioritization of differentially expressed genes and (at the revision stage) the comparison of our data with publicly available gene expression profiles in order to validate the main finding of the paper, that PML knockdown cells have a profile that is similar to differentiated epiblast-like cells.
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Christiana Hadjimichael, Konstantina Chanoumidou, Christoforos Nikolaou, Antonios Klonizakis, Gesthimani-Ioanna Theodosi, Takis Makatounakis, Joseph Papamatheakis, Androniki Kretsovali (2017), Stem Cell Reports, 8(5), 1366-1378
In a nutshell:
Promyelocytic leukemia protein (PML) is a tumor suppressor protein and the major constituent of PML-nuclear bodies, which are known to have a number of regulatory cellular functions, including involvement in programmed cell death, genome stability, antiviral effects and controlling cell division. In this work, we show that PML also plays a major role in the maintenance of pluripotency in mouse ES cells, exhibiting its control on a number of pathways including that of TGF-β at an early stage. The main point of this work is that it provides a link between a protein that has been extensively studied in a different context (its role in cell cycle and apoptosis) and a developmental process such as cell differentiation. Our group's involvement in this work was principally related to the analysis of gene expression data, the identification and prioritization of differentially expressed genes and (at the revision stage) the comparison of our data with publicly available gene expression profiles in order to validate the main finding of the paper, that PML knockdown cells have a profile that is similar to differentiated epiblast-like cells.
ΜicroRNAs for Fine-Tuning of Mouse Embryonic Stem Cell Fate Decision through Regulation of TGF-β Signaling
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Christiana Hadjimichael, Christoforos Nikolaou, Joseph Papamatheakis and Niki Kretsovali, (2016), Stem Cell Reports, 6(3), 292-301
In a nutshell:
Over the past years, microRNAs (miRNAs) have emerged as crucial factors that regulate self-renewal and differentiation of embryonic stem cells (ESCs). Although much is known about their role in maintaining ESC pluripotency, the mechanisms by which they affect cell fate decisions remain poorly understood. By performing deep sequencing to profile miRNA expression in mouse ESCs (mESCs) and differentiated embryoid bodies (EBs), we identified four differentially expressed miRNAs. Our group's part in this work was to define a small subset of four miRs (mir-16-1, mir-191, mir-23a, mir-421) with a potential role in stem cell fate through a combination of a) their significant deregulation between stem cells and differentiated EBs b) their being previously unreported to play a role in the process and c) having a number of protein coding gene targets deregulated upon differentiation at the same stage. Collectively, our findings uncover a regulatory network between the studied miRNAs and both branches of TGF-β/BMP-signaling pathways, revealing their importance for ESC lineage decisions.
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Christiana Hadjimichael, Christoforos Nikolaou, Joseph Papamatheakis and Niki Kretsovali, (2016), Stem Cell Reports, 6(3), 292-301
In a nutshell:
Over the past years, microRNAs (miRNAs) have emerged as crucial factors that regulate self-renewal and differentiation of embryonic stem cells (ESCs). Although much is known about their role in maintaining ESC pluripotency, the mechanisms by which they affect cell fate decisions remain poorly understood. By performing deep sequencing to profile miRNA expression in mouse ESCs (mESCs) and differentiated embryoid bodies (EBs), we identified four differentially expressed miRNAs. Our group's part in this work was to define a small subset of four miRs (mir-16-1, mir-191, mir-23a, mir-421) with a potential role in stem cell fate through a combination of a) their significant deregulation between stem cells and differentiated EBs b) their being previously unreported to play a role in the process and c) having a number of protein coding gene targets deregulated upon differentiation at the same stage. Collectively, our findings uncover a regulatory network between the studied miRNAs and both branches of TGF-β/BMP-signaling pathways, revealing their importance for ESC lineage decisions.

PML's role in breast cancer
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Nikoletta Sachini et al, including Christoforos Nikolaou (2019) Promyelocytic leukemia protein (PML) controls breast cancer cell proliferation by modulating Forkhead transcription factors. Molecular Oncology, 13, 1369-1387.
In a nutshell:
Through a transcriptomic profiling we identified a large number of PML‐deregulated genes associated with various cell processes. Among them, cell cycle‐ and division‐related genes and their cognitive regulators are highly ranked. In this study, we focused on previously unknown PML targets, namely the Forkhead transcription factors. PML suppresses the Forkhead box subclass M1 (FOXM1) transcription factor at both the RNA and protein levels, along with many of its gene targets. We show that FOXM1 interacts with PMLIV primarily via its DNA‐binding domain and dynamically colocalizes in PML nuclear bodies. In parallel, PML modulates the activity of Forkhead box O3 (FOXO3), a factor opposing certain FOXM1 activities, to promote cell survival and stress resistance. We report that inducible PMLIV expression inhibits cell proliferation as well as self‐renewal and impairs cell cycle progression of breast cancer cell lines in a reversible manner.
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Nikoletta Sachini et al, including Christoforos Nikolaou (2019) Promyelocytic leukemia protein (PML) controls breast cancer cell proliferation by modulating Forkhead transcription factors. Molecular Oncology, 13, 1369-1387.
In a nutshell:
Through a transcriptomic profiling we identified a large number of PML‐deregulated genes associated with various cell processes. Among them, cell cycle‐ and division‐related genes and their cognitive regulators are highly ranked. In this study, we focused on previously unknown PML targets, namely the Forkhead transcription factors. PML suppresses the Forkhead box subclass M1 (FOXM1) transcription factor at both the RNA and protein levels, along with many of its gene targets. We show that FOXM1 interacts with PMLIV primarily via its DNA‐binding domain and dynamically colocalizes in PML nuclear bodies. In parallel, PML modulates the activity of Forkhead box O3 (FOXO3), a factor opposing certain FOXM1 activities, to promote cell survival and stress resistance. We report that inducible PMLIV expression inhibits cell proliferation as well as self‐renewal and impairs cell cycle progression of breast cancer cell lines in a reversible manner.

GRG5 as neural stem cell decision switch
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Konstantina Chanoumidou et al including Christoforos Nikolaou (2018) Groucho related gene 5 (GRG5) is involved in embryonic and neural stem cell state decisions. Scientific Reports 2018 Sep 13;8(1):13790.
In a nutshell:
We combine loss and gain of function approaches to demonstrate the pleiotropic roles of the Groucho Related Gene 5 (GRG5) protein in stem cells. By analyzing complete gene expression profiles from: a) the ablation of GRG5 and b) its overexpression we find that the first promotes neuroectodermal specification via Wnt and BMP signaling suppression, while the second leads to enhanced self-renewal and acquisition of cancer cell-like properties. We show this first at genome-wide level through functional analysis of gene expression through the enrichment of specific gene ontology categories and molecular pathways and then with specific targeted experiments against key signaling factors.
(in collaboration with the Kretsovali and Papamatheakis labs, IMBB-FORTH)
Konstantina Chanoumidou et al including Christoforos Nikolaou (2018) Groucho related gene 5 (GRG5) is involved in embryonic and neural stem cell state decisions. Scientific Reports 2018 Sep 13;8(1):13790.
In a nutshell:
We combine loss and gain of function approaches to demonstrate the pleiotropic roles of the Groucho Related Gene 5 (GRG5) protein in stem cells. By analyzing complete gene expression profiles from: a) the ablation of GRG5 and b) its overexpression we find that the first promotes neuroectodermal specification via Wnt and BMP signaling suppression, while the second leads to enhanced self-renewal and acquisition of cancer cell-like properties. We show this first at genome-wide level through functional analysis of gene expression through the enrichment of specific gene ontology categories and molecular pathways and then with specific targeted experiments against key signaling factors.
Circulating MicroRNAs Regulating DNA Damage Response and Responsiveness to Cisplatin in the Prognosis of Patients with Non-Small Cell Lung Cancer Treated with First-Line Platinum Chemotherapy
(in collaboration with the lab of Sofia Aggelaki, University of Crete)
Chara Papadaki, Alexia Monastirioti, Konstantinos Rounis, Dimitrios Makrakis, Konstantinos Kalbakis, Christoforos Nikolaou, Dimitrios Mavroudis and Sofia Agelaki (2020) Cancers, 12(5):1282
In a nutshell:
We studied microRNA expression levels in the plasma of patients with non-small cell lung cancer (NSCLC) obtained prior to initiation of first-line platinum chemotherapy, in order to examine if they can predict the patients' responsiveness to therapy. We found the expression of microRNA (miR)-21, miR-128, miR-155, and miR-181a to be correlated with patients' outcomes. MiR-128, miR-155, and miR-181a expressions were higher in patients compared to healthy donors. High miR-128 and miR-155 were correlated with shorter overall survival (OS), whereas performance status (PS) 2 and high miR-128 independently predicted for decreased OS. Our study showed for the first time that plasma miR-128 and miR-155 hold independent prognostic implications in NSCLC patients treated with platinum-based chemotherapy possibly related to their involvement in tumor response to hypoxia.
(in collaboration with the lab of Sofia Aggelaki, University of Crete)
Chara Papadaki, Alexia Monastirioti, Konstantinos Rounis, Dimitrios Makrakis, Konstantinos Kalbakis, Christoforos Nikolaou, Dimitrios Mavroudis and Sofia Agelaki (2020) Cancers, 12(5):1282
In a nutshell:
We studied microRNA expression levels in the plasma of patients with non-small cell lung cancer (NSCLC) obtained prior to initiation of first-line platinum chemotherapy, in order to examine if they can predict the patients' responsiveness to therapy. We found the expression of microRNA (miR)-21, miR-128, miR-155, and miR-181a to be correlated with patients' outcomes. MiR-128, miR-155, and miR-181a expressions were higher in patients compared to healthy donors. High miR-128 and miR-155 were correlated with shorter overall survival (OS), whereas performance status (PS) 2 and high miR-128 independently predicted for decreased OS. Our study showed for the first time that plasma miR-128 and miR-155 hold independent prognostic implications in NSCLC patients treated with platinum-based chemotherapy possibly related to their involvement in tumor response to hypoxia.

Circulating miRNAs as Potential Biomarkers in Prostate Cancer Patients Undergoing Radiotherapy
(in collaboration with the lab of Sofia Aggelaki, University of Crete)
Stefanos Kachris, Chara Papadaki, Konstantinos Rounis, Eliza Tsitoura, Chrysanthi Kokkinaki, Christoforos Nikolaou, George Sourvinos and Dimitrios Mavroudis. (2022), Cancer Manag Res. 13: 8257–8271
In a nutshell:
Disease recurrence is a major concern in patients with localized prostate cancer (PCa) following treatment with radiotherapy (RT), and few studies have evaluated the clinical relevance of microRNAs (miRNAs) prior and post-RT. We aimed to investigate the significance of miRNAs in the outcomes of prostate cancer patients undergoing radiotherapy and to identify the related pathways through bioinformatics analysis. The expression levels of miR-21, miR-106b, miR-141 and miR-375 involved in the response to radiotherapy were assessed by RT-qPCR in the serum of PCa patients prior- and post-RT. In the whole group of patients, high expression levels of miR-21 prior-RT and of miR-106b post-RT were associated with significantly shorter overall survival. No associations were observed among miR-141 and miR-375 expression levels with clinicopathological features or treatment outcome. Bioinformatics analysis revealed significant enrichment in DNA damage response pathways. Circulating miRNAs prior or post-RT may hold prognostic implications in patients with PCa.
(in collaboration with the lab of Sofia Aggelaki, University of Crete)
Stefanos Kachris, Chara Papadaki, Konstantinos Rounis, Eliza Tsitoura, Chrysanthi Kokkinaki, Christoforos Nikolaou, George Sourvinos and Dimitrios Mavroudis. (2022), Cancer Manag Res. 13: 8257–8271
In a nutshell:
Disease recurrence is a major concern in patients with localized prostate cancer (PCa) following treatment with radiotherapy (RT), and few studies have evaluated the clinical relevance of microRNAs (miRNAs) prior and post-RT. We aimed to investigate the significance of miRNAs in the outcomes of prostate cancer patients undergoing radiotherapy and to identify the related pathways through bioinformatics analysis. The expression levels of miR-21, miR-106b, miR-141 and miR-375 involved in the response to radiotherapy were assessed by RT-qPCR in the serum of PCa patients prior- and post-RT. In the whole group of patients, high expression levels of miR-21 prior-RT and of miR-106b post-RT were associated with significantly shorter overall survival. No associations were observed among miR-141 and miR-375 expression levels with clinicopathological features or treatment outcome. Bioinformatics analysis revealed significant enrichment in DNA damage response pathways. Circulating miRNAs prior or post-RT may hold prognostic implications in patients with PCa.

MicroRNAs Regulating Tumor and Immune Cell Interactions in the Prediction of Relapse in Early Stage Breast Cancer
Chara Papadaki, Konstantina Thomopoulou, Alexia Monastirioti, George Koronakis, Maria A. Papadaki ,Konstantinos Rounis, Lambros Vamvakas, Christoforos Nikolaou, Dimitrios Mavroudis and Sofia Agelaki. (2021) Biomedicines, 9(4), 421
In a nutshell:
MicroRNAs (miRNAs) are involved in the regulation of immune response and hold an important role in tumor immune escape. We investigated the differential expression of the immunomodulatory miR-10b, miR-19a, miR-20a, miR-126, and miR-155 in the plasma of healthy women and patients with early stage breast cancer and interrogated their role in the prediction of patients’ relapse. Blood samples were obtained from healthy women (n = 20) and patients with early stage breast cancer before adjuvant chemotherapy. Relapse predicting models were developed using binary logistic regression and receiver operating curves (ROC) were constructed to determine miRNA sensitivity and specificity. Combined miR-19a and miR-20a expression had the highest performance in discriminating patients with early relapse . Finally, miR-10b in combination with lymph node status and grade had the highest accuracy to discriminate patients with late relapse. The robustness of the relapse predicting models was further confirmed in a 10-fold cross validation. Deregulation of circulating miRNAs involved in tumor-immune interactions may predict relapse in early stage breast cancer. Their successful clinical integration could potentially address the significance challenge of treatment escalation or de-escalation according to the risk of recurrence.
Chara Papadaki, Konstantina Thomopoulou, Alexia Monastirioti, George Koronakis, Maria A. Papadaki ,Konstantinos Rounis, Lambros Vamvakas, Christoforos Nikolaou, Dimitrios Mavroudis and Sofia Agelaki. (2021) Biomedicines, 9(4), 421
In a nutshell:
MicroRNAs (miRNAs) are involved in the regulation of immune response and hold an important role in tumor immune escape. We investigated the differential expression of the immunomodulatory miR-10b, miR-19a, miR-20a, miR-126, and miR-155 in the plasma of healthy women and patients with early stage breast cancer and interrogated their role in the prediction of patients’ relapse. Blood samples were obtained from healthy women (n = 20) and patients with early stage breast cancer before adjuvant chemotherapy. Relapse predicting models were developed using binary logistic regression and receiver operating curves (ROC) were constructed to determine miRNA sensitivity and specificity. Combined miR-19a and miR-20a expression had the highest performance in discriminating patients with early relapse . Finally, miR-10b in combination with lymph node status and grade had the highest accuracy to discriminate patients with late relapse. The robustness of the relapse predicting models was further confirmed in a 10-fold cross validation. Deregulation of circulating miRNAs involved in tumor-immune interactions may predict relapse in early stage breast cancer. Their successful clinical integration could potentially address the significance challenge of treatment escalation or de-escalation according to the risk of recurrence.
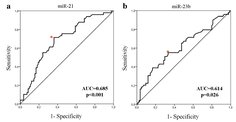
Circulating miRNA and breast cancer
(in collaboration with the lab of Sofia Aggelaki, University of Crete)
Chara Papadaki et al including Christoforos Nikolaou (2018) Circulating microRNAs in the early prediction of disease recurrence in primary breast cancer. Breast Cancer Research. 201820:72
In a nutshell:
Circulating microRNAs are increasingly become a valuable molecular marker for a number of pathological conditions, including cancer. A set of microRNAs that can be detected in patients plasma even after breast cancer-related surgery and treatment were analyzed against in cases of cancer relapse against individuals exceeding median survival times. MicroRNAs were prioritized on the basis of detection levels and were subsequently used to train classification models based on logistic regression and random forests. We found that a combination of three miRs (mir21, mir23b and mir190) was highly predictive of the relapse probability and this was further improved after addition of clinical properties such as lymph node infiltration and tumor grade. Together, through this work we suggest a clear link between circulating microRNAs and the possibility of relapse in breast cancer patients.
(in collaboration with the lab of Sofia Aggelaki, University of Crete)
Chara Papadaki et al including Christoforos Nikolaou (2018) Circulating microRNAs in the early prediction of disease recurrence in primary breast cancer. Breast Cancer Research. 201820:72
In a nutshell:
Circulating microRNAs are increasingly become a valuable molecular marker for a number of pathological conditions, including cancer. A set of microRNAs that can be detected in patients plasma even after breast cancer-related surgery and treatment were analyzed against in cases of cancer relapse against individuals exceeding median survival times. MicroRNAs were prioritized on the basis of detection levels and were subsequently used to train classification models based on logistic regression and random forests. We found that a combination of three miRs (mir21, mir23b and mir190) was highly predictive of the relapse probability and this was further improved after addition of clinical properties such as lymph node infiltration and tumor grade. Together, through this work we suggest a clear link between circulating microRNAs and the possibility of relapse in breast cancer patients.
On sequence analysis

Revisiting the Non-Coding Nature of Pospiviroids
Konstantina Katsarou, Charith Raj Adkar-Purushothama, Emilios Tassios, Martina Samiotaki, Christos Andronis, Purificación Lisón, Christoforos Nikolaou, Jean-Pierre Perreault and Kriton Kalantidis. (2022) Cells 2022, 11(2), 265
In a nutshell:
Viroids are small, circular, highly structured pathogens that infect a broad range of plants, causing economic losses. Since their discovery in the 1970s, they have been considered as non-coding pathogens. In the last few years, the discovery of other RNA entities, similar in terms of size and structure, that were shown to be translated (e.g., cirRNAs, precursors of miRNA, RNA satellites) as well as studies showing that some viroids are located in ribosomes, have reignited the idea that viroids may be translated. In this study, we used advanced bioinformatic analysis, in vitro experiments and LC-MS/MS to search for small viroid peptides of the PSTVd. Our results suggest that in our experimental conditions, even though the circular form of PSTVd is found in ribosomes, no produced peptides were identified. This indicates that the presence of PSTVd in ribosomes is most probably not related to peptide production but rather to another unknown function that requires further study.
Konstantina Katsarou, Charith Raj Adkar-Purushothama, Emilios Tassios, Martina Samiotaki, Christos Andronis, Purificación Lisón, Christoforos Nikolaou, Jean-Pierre Perreault and Kriton Kalantidis. (2022) Cells 2022, 11(2), 265
In a nutshell:
Viroids are small, circular, highly structured pathogens that infect a broad range of plants, causing economic losses. Since their discovery in the 1970s, they have been considered as non-coding pathogens. In the last few years, the discovery of other RNA entities, similar in terms of size and structure, that were shown to be translated (e.g., cirRNAs, precursors of miRNA, RNA satellites) as well as studies showing that some viroids are located in ribosomes, have reignited the idea that viroids may be translated. In this study, we used advanced bioinformatic analysis, in vitro experiments and LC-MS/MS to search for small viroid peptides of the PSTVd. Our results suggest that in our experimental conditions, even though the circular form of PSTVd is found in ribosomes, no produced peptides were identified. This indicates that the presence of PSTVd in ribosomes is most probably not related to peptide production but rather to another unknown function that requires further study.
Menzerath-Altmann law in mammalian exons
Christoforos Nikolaou (2014) Computational Biology and Chemistry. 53.
In a nutshell:
According to the Menzerath-Altmann law of statistical linguistics, the average length of a word's syllables is smaller as their number increases. An analogy of this law in molecular biology has already been pointed out by Wentian Li (see here). Li has shown that genes with a great number of exons also tend to have smaller exons. In this work we expand his finding by investigating the link between this property and the evolutionary dymamics of exons. In particular we focus the following question: Are conserved exons more prone to follow the Menzerath-Altmann law than non-conserved ones? By a detailed analysis of the complete set of mouse genes we show that less conserved exons are more likely responsible for the Menzerath-Altmann-like behaviour and that as the sequence constraints (i.e. the requirement that exons conserve their sequence) increases the concordance with the law diminishes. The implications of our findings are that the existence of this law is mostly due to the dynamics of exon birth and that as the exons are progressively attributed with more important functions and genes become more complex (with alternative transcript isoforms) this property is gradually lost. The linguistics equivalent (although quite hard to demonstrate) would be to expect that the law will not hold for words with multiple meanings or words that have become too important in the language such as the ones used more frequently.
Christoforos Nikolaou (2014) Computational Biology and Chemistry. 53.
In a nutshell:
According to the Menzerath-Altmann law of statistical linguistics, the average length of a word's syllables is smaller as their number increases. An analogy of this law in molecular biology has already been pointed out by Wentian Li (see here). Li has shown that genes with a great number of exons also tend to have smaller exons. In this work we expand his finding by investigating the link between this property and the evolutionary dymamics of exons. In particular we focus the following question: Are conserved exons more prone to follow the Menzerath-Altmann law than non-conserved ones? By a detailed analysis of the complete set of mouse genes we show that less conserved exons are more likely responsible for the Menzerath-Altmann-like behaviour and that as the sequence constraints (i.e. the requirement that exons conserve their sequence) increases the concordance with the law diminishes. The implications of our findings are that the existence of this law is mostly due to the dynamics of exon birth and that as the exons are progressively attributed with more important functions and genes become more complex (with alternative transcript isoforms) this property is gradually lost. The linguistics equivalent (although quite hard to demonstrate) would be to expect that the law will not hold for words with multiple meanings or words that have become too important in the language such as the ones used more frequently.
A model to explain the evolution of homo-polymeric tracts in genomic sequences
Christoforos Nikolaou and Yannis Almirantis (2005) 'Word' preference in the genomic text and genome evolution. Different modes of n-tuplet usage in coding and noncoding sequences. Journal of Molecular Evolution, 61, 23-35
In a nutshell:
In genomic sequences, especially the ones of higher eukaryotes, omo-polymeric tracts, (sequences with many of the same or similar nucleotides in a row, such as AAAAAA, TTTTT, GCGGCCCGG) are much more common in the non-coding part of the genome, occurring away from the genes. In this work we propose a plausible evolutionary scenario to explain this phenomenon. According to our model a process called DNA-strand slippage may account for this discrepancy. During replication of the DNA, the two strands may slide (slip) against each other and so errors occur that are propagated to the next generation. Slippage of the strands are much more frequent in the case of homo-polymeric tracts as the principle of complementarity is not disturbed in this case. Moreover, slippage events result in a further increase of the size of homo-polymeric tracts. In this sense we have a synergistic effect with more and longer homo-polymeric tracts leading their own expansion (which is only hindered by selective processes when it comes to disrupt functional elements of the genome).
Christoforos Nikolaou and Yannis Almirantis (2005) 'Word' preference in the genomic text and genome evolution. Different modes of n-tuplet usage in coding and noncoding sequences. Journal of Molecular Evolution, 61, 23-35
In a nutshell:
In genomic sequences, especially the ones of higher eukaryotes, omo-polymeric tracts, (sequences with many of the same or similar nucleotides in a row, such as AAAAAA, TTTTT, GCGGCCCGG) are much more common in the non-coding part of the genome, occurring away from the genes. In this work we propose a plausible evolutionary scenario to explain this phenomenon. According to our model a process called DNA-strand slippage may account for this discrepancy. During replication of the DNA, the two strands may slide (slip) against each other and so errors occur that are propagated to the next generation. Slippage of the strands are much more frequent in the case of homo-polymeric tracts as the principle of complementarity is not disturbed in this case. Moreover, slippage events result in a further increase of the size of homo-polymeric tracts. In this sense we have a synergistic effect with more and longer homo-polymeric tracts leading their own expansion (which is only hindered by selective processes when it comes to disrupt functional elements of the genome).
Power-laws in orphan and gene related CpG Islands
(in collaboration with Yannis Almirantis lab, NCSR "Demokritos")
Giannis Tsiagkas, Christoforos Nikolaou and Yannis Almirantis Y (2014) Computational Biology and Chemistry.
In a nutshell:
CpG islands are regions of extreme nucleotide composition in eukarytotic genomes, whose established role in regulation has been extensively studied. With a mean size of 200bp, very high percentage in guanine and cytosine basis and an elevated preference for the CpG dinucleotide that forms the main substrate for DNA methyl-transferases, the great majority of CpG islands are found to be methylated in somatic cells. Their regulatory function is exerted predominantly when they remain unmethylated and in close proximity to transcription start sites of genes, whose expression they are activating. Nonetheless, a non-negligible subset of higher eukaryotic CpG islands are found to be located away from known genes and their lack of attribution to a gene locus has earned them their term "orphans". In what sense are these "orphan" CpG islands different than the ones located close to known genes? In this work, a collaboration with the group of Yannis Almirantis at the NCSR "Demokritos", we examined one particular property that is indirectly related with the assumed evolutionary history of genomic components, namely their genomic localization in the form of distributions of their distances. We found that both "orphan" and "gene-associated" CpG islands exhibit the same overall properties in terms of genome localization and, moreover, that their distribution in chromosomes of a great number of eukaryotes fits well with an evolutionary scenario of duplication coupled with extensive elimination of genomic elements.
(in collaboration with Yannis Almirantis lab, NCSR "Demokritos")
Giannis Tsiagkas, Christoforos Nikolaou and Yannis Almirantis Y (2014) Computational Biology and Chemistry.
In a nutshell:
CpG islands are regions of extreme nucleotide composition in eukarytotic genomes, whose established role in regulation has been extensively studied. With a mean size of 200bp, very high percentage in guanine and cytosine basis and an elevated preference for the CpG dinucleotide that forms the main substrate for DNA methyl-transferases, the great majority of CpG islands are found to be methylated in somatic cells. Their regulatory function is exerted predominantly when they remain unmethylated and in close proximity to transcription start sites of genes, whose expression they are activating. Nonetheless, a non-negligible subset of higher eukaryotic CpG islands are found to be located away from known genes and their lack of attribution to a gene locus has earned them their term "orphans". In what sense are these "orphan" CpG islands different than the ones located close to known genes? In this work, a collaboration with the group of Yannis Almirantis at the NCSR "Demokritos", we examined one particular property that is indirectly related with the assumed evolutionary history of genomic components, namely their genomic localization in the form of distributions of their distances. We found that both "orphan" and "gene-associated" CpG islands exhibit the same overall properties in terms of genome localization and, moreover, that their distribution in chromosomes of a great number of eukaryotes fits well with an evolutionary scenario of duplication coupled with extensive elimination of genomic elements.
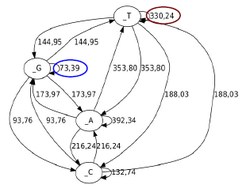
N-gram Graphs in Genomic Sequences.
(in collaboration with George Paliouras lab, NCSR "Demokritos")
Dimitris Polychronopoulos et al including Christoforos Nikolaou (2014) Analysis and Classification of Constrained DNA Elements with N-gram Graphs and Genomic Signatures, Algorithms for Computational Biology, Lecture Notes in Computer Science, 220-234
In a nutshell:
When looking at a genomic sequence one is faced with a long series of characters from the 4-letter alphabet (A,G,C and T). Discerning the underlying order of the message(s) contained in these strings of characters is a problem that has been up to now only partially solved with existent techniques. Most of those treat the series (the DNA text) as a bag of oligonucleotide "words" trying to assess their relative frequencies. In this work, we use a novel methodology, that takes into account not only the number each nucleotide k-mer occurs in a sequence but also the degree to which it is found within the vicinity of others. In this sense we construct networks of "words", through N-gram Graphs (NGG) and use them to characterize a very particular "obscure" category of DNA sequences that are extremely conserved without yet having a known function (conserved non-coding elements or CNE). These sequences, we found, have distinct compositional characteristics that distinguish them from conserved coding sequences (such as exons) in the same organism. The work is the result of an ongoing collaboration with the Software and Knowledge Engineering Laboratory (SKEL) at NCSR "Demokritos" and builds upon the concept of N-gram graphs as introduced in the PhD thesis of our friend and collaborator George Giannakopoulos.
(in collaboration with George Paliouras lab, NCSR "Demokritos")
Dimitris Polychronopoulos et al including Christoforos Nikolaou (2014) Analysis and Classification of Constrained DNA Elements with N-gram Graphs and Genomic Signatures, Algorithms for Computational Biology, Lecture Notes in Computer Science, 220-234
In a nutshell:
When looking at a genomic sequence one is faced with a long series of characters from the 4-letter alphabet (A,G,C and T). Discerning the underlying order of the message(s) contained in these strings of characters is a problem that has been up to now only partially solved with existent techniques. Most of those treat the series (the DNA text) as a bag of oligonucleotide "words" trying to assess their relative frequencies. In this work, we use a novel methodology, that takes into account not only the number each nucleotide k-mer occurs in a sequence but also the degree to which it is found within the vicinity of others. In this sense we construct networks of "words", through N-gram Graphs (NGG) and use them to characterize a very particular "obscure" category of DNA sequences that are extremely conserved without yet having a known function (conserved non-coding elements or CNE). These sequences, we found, have distinct compositional characteristics that distinguish them from conserved coding sequences (such as exons) in the same organism. The work is the result of an ongoing collaboration with the Software and Knowledge Engineering Laboratory (SKEL) at NCSR "Demokritos" and builds upon the concept of N-gram graphs as introduced in the PhD thesis of our friend and collaborator George Giannakopoulos.
Improving GeneFinding Tools
Yannis Almirantis and Christoforos Nikolaou (2005) Multi-criterial Coding Sequence Prediction. Combination of GeneMark with two novel, coding character-specific quantities. Computers in Biology and Medicine, 35, 627-643
In a nutshell:
Gene finding of a benchmark program (GeneMark) is improved through the use of additional information at the small (2-4 nucleotides) and the middle (10-100 nucleotide) scales. We have applied a "patch" to GeneMark consisting of two independently developed gene-potential measuring methods to show that their implementation on top of GeneMark's predictions increases its overall performance. In brief, we show how ensembles of methods can actually make more efficient predictions in terms of gene finding.
Yannis Almirantis and Christoforos Nikolaou (2005) Multi-criterial Coding Sequence Prediction. Combination of GeneMark with two novel, coding character-specific quantities. Computers in Biology and Medicine, 35, 627-643
In a nutshell:
Gene finding of a benchmark program (GeneMark) is improved through the use of additional information at the small (2-4 nucleotides) and the middle (10-100 nucleotide) scales. We have applied a "patch" to GeneMark consisting of two independently developed gene-potential measuring methods to show that their implementation on top of GeneMark's predictions increases its overall performance. In brief, we show how ensembles of methods can actually make more efficient predictions in terms of gene finding.

Measures of oligonucleotide Assymmetry
Christoforos Nikolaou and Yannis Almirantis (2003) Mutually symmetric and complementary triplets: Differences in their use distinguish systematically between coding and non-coding genomic sequences. Journal of Theoretical Biology, 223, 477-487
Christoforos Nikolaou and Yannis Almirantis (2004) Measuring the Coding Potential of Genomic Sequences through a combination of Triplet Occurrence Patterns and RNY Preference. Journal of Molecular Evolution, 59, 309-316
In a nutshell:
In natural languages palindromes (mirror-image words or phrases) such as "Able:Elba" or "Madam, I 'm Adam" are very rare. Moreover it is natural that their two components do not occur with similar frequencies (Able is much more common than Elba) as a direct result of the codification process. What happens in genomic DNA sequences? In these two papers we have addressed the problem of symmetry in trinucleotide occurrences in coding and non-coding sequences and have verified that palindrome sequences occur with rather distinct frequencies only in the first, that is in the part of the genome where coding constraints are more prominent. Moreover we show that two measures designed to quantify the discrepancy between symmetrical (palindrome) trinucleotides (having accounted for background sequence composition) are suitable for the efficient classification of genomic sequences into coding and non-coding.
Christoforos Nikolaou and Yannis Almirantis (2003) Mutually symmetric and complementary triplets: Differences in their use distinguish systematically between coding and non-coding genomic sequences. Journal of Theoretical Biology, 223, 477-487
Christoforos Nikolaou and Yannis Almirantis (2004) Measuring the Coding Potential of Genomic Sequences through a combination of Triplet Occurrence Patterns and RNY Preference. Journal of Molecular Evolution, 59, 309-316
In a nutshell:
In natural languages palindromes (mirror-image words or phrases) such as "Able:Elba" or "Madam, I 'm Adam" are very rare. Moreover it is natural that their two components do not occur with similar frequencies (Able is much more common than Elba) as a direct result of the codification process. What happens in genomic DNA sequences? In these two papers we have addressed the problem of symmetry in trinucleotide occurrences in coding and non-coding sequences and have verified that palindrome sequences occur with rather distinct frequencies only in the first, that is in the part of the genome where coding constraints are more prominent. Moreover we show that two measures designed to quantify the discrepancy between symmetrical (palindrome) trinucleotides (having accounted for background sequence composition) are suitable for the efficient classification of genomic sequences into coding and non-coding.
Modified Standard Deviation (MSD)
Christoforos Nikolaou and Yannis Almirantis (2002) A study of the middle-scale nucleotide clustering in DNA sequences of various origin and functionality, by means of a method based on a Modified Standard Deviation. Journal of Theoretical Biology, 217, 479-492
In a nutshell:
DNA sequences are non-random in what concerns the juxtaposition of nucleotides. We here present a measure for the quantification of this non-randomness addressing the middle-scale (the length order of around 10-100 nucleotides) to show that through a carefully designed quality destined to measure the clustering of similar nucleotides one can effectively distinguish between coding and non-coding sequences.
Christoforos Nikolaou and Yannis Almirantis (2002) A study of the middle-scale nucleotide clustering in DNA sequences of various origin and functionality, by means of a method based on a Modified Standard Deviation. Journal of Theoretical Biology, 217, 479-492
In a nutshell:
DNA sequences are non-random in what concerns the juxtaposition of nucleotides. We here present a measure for the quantification of this non-randomness addressing the middle-scale (the length order of around 10-100 nucleotides) to show that through a carefully designed quality destined to measure the clustering of similar nucleotides one can effectively distinguish between coding and non-coding sequences.
Data Analysis on various projects
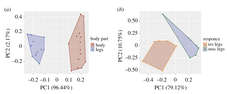
Data Analysis on proteomic data from an insecticide resistance study
(in collaboration with John Vontas lab, AUA)
Vasileia Balabanidou et al, including Christoforos Nikolaou and John Vontas (2019) Mosquitoes cloak their legs to resist insecticides, Proceedings of the Royal Society B, 286, 20191091
In a nutshell:
By comparing legs of mosquitoes, the most relevant insect tissue for insecticide uptake, we show that resistant mosquitoes largely remodel their leg cuticles via enhanced deposition of cuticular proteins and chitin, corroborating a leg-thickening phenotype. Moreover, we show that resistant female mosquitoes seal their leg cuticles with higher total and different relative amounts of cuticular hydrocarbons, compared with susceptible ones. The structural and functional alterations in Anopheles female mosquito legs are associated with a reduced uptake of insecticides, substantially contributing to the resistance phenotype.
(in collaboration with John Vontas lab, AUA)
Vasileia Balabanidou et al, including Christoforos Nikolaou and John Vontas (2019) Mosquitoes cloak their legs to resist insecticides, Proceedings of the Royal Society B, 286, 20191091
In a nutshell:
By comparing legs of mosquitoes, the most relevant insect tissue for insecticide uptake, we show that resistant mosquitoes largely remodel their leg cuticles via enhanced deposition of cuticular proteins and chitin, corroborating a leg-thickening phenotype. Moreover, we show that resistant female mosquitoes seal their leg cuticles with higher total and different relative amounts of cuticular hydrocarbons, compared with susceptible ones. The structural and functional alterations in Anopheles female mosquito legs are associated with a reduced uptake of insecticides, substantially contributing to the resistance phenotype.
