A recent advanced online publication in Nature (Lappalainen et al, Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013 Sep 15) features the first concise attempt to assess transcriptome variation in the human genome. This forms part of the Geuvadis consortium which brings together scientists from various research centers in Europe and the US under the coordination of X. Estivill and R. Guigo from CRG, Barcelona.
The data used: Primary lymphoblastoid cells of 462 individuals of various ethnic backgrounds were analyzed for total mRNA and miRNA levels by applying high-throughput RNASeq in different platforms and in different scientific institutes. 452 out of those were also genotyped as part of the human 1000 genomes project. To cut the long story short this is a huge amount of data at both genome and transcriptome level.
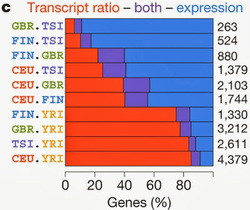
The analysis: The analysis doesn't involve really complex approach but is made extremely complicated from the sheer amount of data analyzed. Two of the main goals of the Geuvadis consortium are precisely to design efficient manipulation and storage techniques for big data and to test the robustness and replicability of transcriptome analysis. Both objectives were met. On the other hand very few novel or unexpected conclusion were drawn from this study. Yes, there is variation in transcriptome, and yes for the most part it is regulatory meaning that differences in gene expression are not qualitative but quantitative. This is not unexpected. On the other hand one interesting finding, that variation takes place mostly at the relative transcript ratio of the same genes instead of gene expression levels (depicted in the Figure) is not very straight-forward and will probably prove extremely difficult to further validate.
What's next: Let me guess; EVEN more sequencing about to come.
Read more: On the assessment of RNASeq variability and whether this is an efficient method to accurately reflect transcription levels in human (spoiler alert: It is!) (t' Hoen et al., 2013) and on the 1000 genomes project assessing the genomic variation in the human population (Mc Vean et al., 2012)
The data used: Primary lymphoblastoid cells of 462 individuals of various ethnic backgrounds were analyzed for total mRNA and miRNA levels by applying high-throughput RNASeq in different platforms and in different scientific institutes. 452 out of those were also genotyped as part of the human 1000 genomes project. To cut the long story short this is a huge amount of data at both genome and transcriptome level.
The analysis: The analysis doesn't involve really complex approach but is made extremely complicated from the sheer amount of data analyzed. Two of the main goals of the Geuvadis consortium are precisely to design efficient manipulation and storage techniques for big data and to test the robustness and replicability of transcriptome analysis. Both objectives were met. On the other hand very few novel or unexpected conclusion were drawn from this study. Yes, there is variation in transcriptome, and yes for the most part it is regulatory meaning that differences in gene expression are not qualitative but quantitative. This is not unexpected. On the other hand one interesting finding, that variation takes place mostly at the relative transcript ratio of the same genes instead of gene expression levels (depicted in the Figure) is not very straight-forward and will probably prove extremely difficult to further validate.
What's next: Let me guess; EVEN more sequencing about to come.
Read more: On the assessment of RNASeq variability and whether this is an efficient method to accurately reflect transcription levels in human (spoiler alert: It is!) (t' Hoen et al., 2013) and on the 1000 genomes project assessing the genomic variation in the human population (Mc Vean et al., 2012)
 RSS Feed
RSS Feed
