Our group's last paper has just appeared in Nucleic Acids Research. In it we introduce a new concept that is related to the architecture of eukaryotic genomes and how it may have evolved to segregate functions in different genomic compartments in a way that is reminiscent of the semi-spontaneous social/ratial segregation of cities.
Genome Urbanization is inspired by the pioneering works on social segregation by Thomas Schelling whereby weak constraints may aggregate to shape macroscopic behaviour in various systems. Using an already published experiment in which we had previously studied how the chromatin structure of yeast promoters may shape gene expression under the accumulation of topological stress, we set out to investigate whether differential gene expression upon a structurally-related stimulus may be reflected upon the spatial organization of genes. The first thing we found was that when put under topological stress, genes tend to form clusters in space, with groups of 6 or more adjacent genes being consistently up- or down-regulated. This was not so much of a surprise. DNA torsional stress acts exactly on the topology of the nucleus and so it would be expected for genes to follow this constraint. Surprisingly though, some clustering of gene expression also happens under other stress conditions such as heat shock or nutrient deprivation albeit to a much lesser extent. What this pointed to is that genes occupy positions in the nucleus (even in lthe inear dimension) that allows them to respond to stimuli in a coordinated manner, thus the "micro-motives" of Schelling may be reflected on the local prerequisites of certain genes to be close to others due to their shared affinities for the same transcription factors or because the local environment is more favourable to their dedicated function.
Gene clustering is, of course, nothing new and many people have spotted preferences for genes that are spatially related in terms of expression, co-evolution or co-regulation. Our work, in this respect, focused on the chromatin and local gene structure of the observed gene clusters. We were thus able to define two distinct genome components with properties so different that the allusion to urban neighborhoods was almost spontaneous.
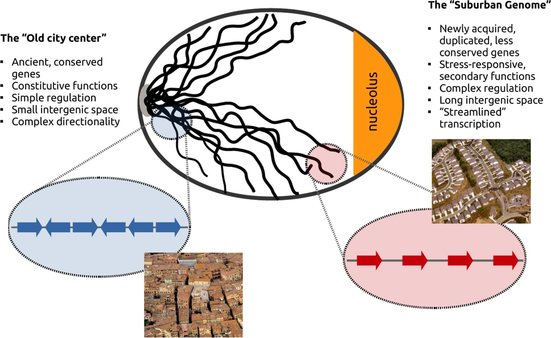
Genes that are "shut-down" upon the accumulation of topological stress were predominantly found close to the centromeres and the nuclear core. They were "old" genes in the sense that they coded for conserved, fundamental functions such as gene transcription and protein processing. More importantly, they were placed within very short distances from each other and with a rather "crammed" orientation that put, very frequently, adjacent genes transcribed in opposing directions. On the other hand, genes that were positively regulated, were found on the other extreme of the nucleus, close to subtelomeric regions. They coded for "newly" acquired stress-response functions, they had complex regulation and were surprisingly aligned with a clear tendency for co-directionality. They also had significantly longer "breathing" intergenic space between them. The set of these structural properties may allow them not only to be "resistant" to topological stress but to even harness DNA supercoiling in order to propel transcription.
This discrepancy in so many levels lead us to propose the Genome Urbanization model according to which, older, more conserved genes are preferentially located in the "old city center". The genome's core resembles the urban plan of a medieval city with its narrow meandering streets leaving little space between houses that appear as if touching each other. At the edges of the chromosomes lies what we call the "suburban genome" where the genome's "nouveau riches", new genes with complex functions that are not necessarily constitutively expressed and are employed only under specific conditions, have created a much different landscape. Here, genes are organized in tandem and with longer intergenic spacers inbetween them, in a way that brings to mind the tract housing of US city suburbia.
Genome Urbanization is inspired by the pioneering works on social segregation by Thomas Schelling whereby weak constraints may aggregate to shape macroscopic behaviour in various systems. Using an already published experiment in which we had previously studied how the chromatin structure of yeast promoters may shape gene expression under the accumulation of topological stress, we set out to investigate whether differential gene expression upon a structurally-related stimulus may be reflected upon the spatial organization of genes. The first thing we found was that when put under topological stress, genes tend to form clusters in space, with groups of 6 or more adjacent genes being consistently up- or down-regulated. This was not so much of a surprise. DNA torsional stress acts exactly on the topology of the nucleus and so it would be expected for genes to follow this constraint. Surprisingly though, some clustering of gene expression also happens under other stress conditions such as heat shock or nutrient deprivation albeit to a much lesser extent. What this pointed to is that genes occupy positions in the nucleus (even in lthe inear dimension) that allows them to respond to stimuli in a coordinated manner, thus the "micro-motives" of Schelling may be reflected on the local prerequisites of certain genes to be close to others due to their shared affinities for the same transcription factors or because the local environment is more favourable to their dedicated function.
Gene clustering is, of course, nothing new and many people have spotted preferences for genes that are spatially related in terms of expression, co-evolution or co-regulation. Our work, in this respect, focused on the chromatin and local gene structure of the observed gene clusters. We were thus able to define two distinct genome components with properties so different that the allusion to urban neighborhoods was almost spontaneous.
Genes that are "shut-down" upon the accumulation of topological stress were predominantly found close to the centromeres and the nuclear core. They were "old" genes in the sense that they coded for conserved, fundamental functions such as gene transcription and protein processing. More importantly, they were placed within very short distances from each other and with a rather "crammed" orientation that put, very frequently, adjacent genes transcribed in opposing directions. On the other hand, genes that were positively regulated, were found on the other extreme of the nucleus, close to subtelomeric regions. They coded for "newly" acquired stress-response functions, they had complex regulation and were surprisingly aligned with a clear tendency for co-directionality. They also had significantly longer "breathing" intergenic space between them. The set of these structural properties may allow them not only to be "resistant" to topological stress but to even harness DNA supercoiling in order to propel transcription.
This discrepancy in so many levels lead us to propose the Genome Urbanization model according to which, older, more conserved genes are preferentially located in the "old city center". The genome's core resembles the urban plan of a medieval city with its narrow meandering streets leaving little space between houses that appear as if touching each other. At the edges of the chromosomes lies what we call the "suburban genome" where the genome's "nouveau riches", new genes with complex functions that are not necessarily constitutively expressed and are employed only under specific conditions, have created a much different landscape. Here, genes are organized in tandem and with longer intergenic spacers inbetween them, in a way that brings to mind the tract housing of US city suburbia.
Genome Urbanization may not be a particular property of the yeast genome, even though unicellularity and the increased gene density compared to more complex eukaryotic genomes is likely to make positional constraints more apparent. We nonetheless believe that similar tendencies may exist in bigger, mammalian genomes and that they may reflect even more intricate regulatory patterns that balance transcriptional homeostasis with expression noise and with the ability to respond to a great number of external and internal stimuli.
Given that the original data were already in our hands since 2006 and the fact that I first presented this concept more than 2 years ago in a talk at the IMBB, FORTH in Crete, this work has been no easy task to complete. It took the combined work of two undergraduate students (Maria Tsochatzidou and Maria Malliarou), the crucial assistance of a fellow bioinformatician (Nikolas Papanikolaou) and the support of long-standing collaborator Joaquim Roca at the CSIC, Barcelona who first introduced me to DNA topology. We are currently looking into many interesting perspectives that this work opens up regarding the evolution of genome architecture in eukaryotes and how gene positioning and chromosome structure may provide insight on the way cells employ transcriptional regulation under various conditions.
This paper is also, strictly speaking, the first paper to come entirely out of our group and thus seeing it published on my son's 5th birthday adds to a sense of accomplishment.
Given that the original data were already in our hands since 2006 and the fact that I first presented this concept more than 2 years ago in a talk at the IMBB, FORTH in Crete, this work has been no easy task to complete. It took the combined work of two undergraduate students (Maria Tsochatzidou and Maria Malliarou), the crucial assistance of a fellow bioinformatician (Nikolas Papanikolaou) and the support of long-standing collaborator Joaquim Roca at the CSIC, Barcelona who first introduced me to DNA topology. We are currently looking into many interesting perspectives that this work opens up regarding the evolution of genome architecture in eukaryotes and how gene positioning and chromosome structure may provide insight on the way cells employ transcriptional regulation under various conditions.
This paper is also, strictly speaking, the first paper to come entirely out of our group and thus seeing it published on my son's 5th birthday adds to a sense of accomplishment.
 RSS Feed
RSS Feed
